
Latest recommendations
| Id | Title * | Authors * | Abstract * | Picture * | Thematic fields * | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
27 May 2025

POSTPRINT
Scalable, accessible and reproducible reference genome assembly and evaluation in GalaxyA new Galaxy workflow to generate and evaluate reference genome assembliesRecommended by Alba Marino , Anna-Sophie Fiston-Lavier and Capucine Mayoud , Anna-Sophie Fiston-Lavier and Capucine Mayoud
Alba Marino (1), Capucine Mayoud (1), Anna-Sophie Fiston-Lavier (1,2) (1) ISEM, Univ Montpellier, CNRS, IRD, Montpellier, France (2) Institut Universitaire de France Biodiversity is the bedrock of many ecosystem services fundamental to human society. Acquiring genome-level information appears increasingly important for a deeper understanding of biodiversity and to plan conservation actions for endangered species (Lewin et al. 2022). Consortia such as the Vertebrate Genomes Project (VGP; Rhie et al. 2021) and the European Reference Genome Atlas (ERGA; Formenti et al. 2022) have been undertaken to coordinate global efforts toward sequencing of all the existing vertebrate and European eukaryotic species, respectively. Indeed, generating genome-scale data across such a wide taxonomic range presents significant challenges—not least the development and long-term maintenance of computational tools and workflows that ensure both reproducibility and transparency. Galaxy offers a user-friendly, web-based environment for executing complex pipelines in a reproducible way, as well as servers for data storage (Bray & Maier 2023). In this context, Larivière et al. (2024) present a major enhancement to reference genome assembly with the development of a scalable, accessible, and reproducible pipeline embedded within the Galaxy platform. The framework has been designed to democratize the production of high-quality genomes, in line with initiatives such as the Earth BioGenome Project (Lewin et al. 2022). It integrates six main stages, namely (1) k-mer genome profiling, (2) phased assembly construction, (3) artefactual duplication purging, (4) scaffolding, (5) decontamination, and (6) mitogenome assembly. The pipeline builds on the expertise of VGP (Rhie et al. 2021) and ERGA (Formenti et al. 2022), while incorporating recent advances in high-fidelity long-read sequencing technologies. A key strength of the pipeline lies in the open availability and its modularity, which enables end-to-end processing from raw reads to curated assemblies while emphasizing reproducibility, transparency, and ease of use (Afgan et al. 2018). Another major advantage is the integration of quality control steps throughout the pipeline. Moreover, the system is designed to accommodate a wide range of input data types and is applicable to a broad spectrum of species (Larivière et al. 2024). Several public Galaxy instances are available worldwide (e.g. in the USA: https://usegalaxy.org; in Europe: https://usegalaxy.eu; in Australia: https://usegalaxy.org.au). These platforms provide free access to computing resources for running complex workflows and analysing large datasets. Nonetheless, certain steps in genome assembly may require more memory (RAM) or processing power (CPU) than the instances can offer, thus demanding access to high-performance computing (HPC) environments. Although cloud execution is mentioned as a means of processing large amounts of data, the manuscript offers little detail on deployment costs or potential technical barriers. Beyond technical and financial considerations, the environmental impact of scaling up genome sequencing and assembly also deserves attention. As more projects are launched and reliance on cloud infrastructure increases, the demand for computing, data storage, and long-term archival will increase substantially. Such operations are energy-intensive and contribute significantly to the environmental footprint of computational biology (Lannelongue & Inouye 2023). While Larivière et al. (2024) rightly emphasize accessibility and scalability, the community must also consider sustainability strategies to limit the ecological impact of large-scale genome initiatives. The authors suggest that the pipeline can be adapted for non-vertebrate species, such as plants or fungi, by adjusting a few parameters (e.g. BUSCO clade selection). However, the pipeline has so far only been validated on vertebrate genomes. Its robustness across taxa with complex genomic features, such as extreme GC content, polyploidy, or high repeat density, will require further benchmarking. Finally, another challenge is keeping the pipeline up to date. The rapid evolution of genome assembly tools (Nurk et al. 2022) contrasts with the often slower update cycles of Galaxy workflows, raising concerns about maintaining best practice standards without active long-term governance. The pipeline would benefit from an additional step to compare the established Galaxy pipeline with new assembly tools better suited to data generating using the latest technologies. In conclusion, Larivière et al. (2024) offer a vital step forward in making reference-quality genome assembly broadly accessible. It is now in the hands of the community to address the remaining open challenges, such as computational accessibility, broader taxonomic validation, environmental sustainability, and further proofing of the pipeline. Afgan E, Baker D, Batut B, van den Beek M, Bouvier D, Čech M, Chilton J, Clements D, Coraor N, Grüning BA, Guerler A, Hillman-Jackson J, Hiltemann S, Jalili V, Rasche H, Soranzo N, Goecks J, Taylor J, Nekrutenko A, Blankenberg D (2018) The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Research, 46, W537–W544. https://doi.org/10.1093/nar/gky379 Bray S, Maier W. (2023) Automating Galaxy workflows using the command line. Galaxy Training Network. https://training.galaxyproject.org/training-material/topics/galaxy-interface/tutorials/workflow-automation/tutorial.html Formenti G, Theissinger K, Fernandes C, Bista I, Bombarely A, Bleidorn C, et al. (2022) The era of reference genomes in conservation genomics. Trends in Ecology & Evolution, 37, 197–202. https://doi.org/10.1016/j.tree.2021.11.008 Lannelongue, L, Inouye, M (2023) Carbon footprint estimation for computational research. Nat Rev Methods Primers 3, 9. https://doi.org/10.1038/s43586-023-00202-5 Larivière D, Abueg L, Brajuka N, Gallardo-Alba C, Grüning B, Ko BJ, Ostrovsky A, Palmada-Flores M, Pickett BD, Rabbani K, Antunes A, Balacco JR, Chaisson MJP, Cheng H, Collins J, Couture M, Denisova A, Fedrigo O, Gallo GR, Giani AM, Gooder GM, Horan K, Jain N, Johnson C, Kim H, Lee C, Marques-Bonet T, O’Toole B, Rhie A, Secomandi S, Sozzoni M, Tilley T, Uliano-Silva M, van den Beek M, Williams RW, Waterhouse RM, Phillippy AM, Jarvis ED, Schatz MC, Nekrutenko A, Formenti G (2024) Scalable, accessible and reproducible reference genome assembly and evaluation in Galaxy. Nature Biotechnology, 42, 367–370. https://doi.org/10.1038/s41587-023-02100-3 Lewin HA, Richards S, Lieberman Aiden E, Allende ML, et al. (2022) The Earth BioGenome Project 2020: Starting the clock. Proceedings of the National Academy of Sciences, 119, e2115635118. https://doi.org/10.1073/pnas.2115635118 Nurk S, Koren S, Rhie A, Rautiainen M, Bzikadze AV, et al. (2022) The complete sequence of a human genome. Science, 376, 44–53. https://doi.org/10.1126/science.abj6987 Rhie A, McCarthy SA, Fedrigo O, Damas J, Formenti G, et al. (2021) Towards complete and error-free genome assemblies of all vertebrate species. Nature, 592, 737–746. https://doi.org/10.1038/s41586-021-03451-0 | Scalable, accessible and reproducible reference genome assembly and evaluation in Galaxy | Delphine Larivière, Linelle Abueg, Nadolina Brajuka, Cristóbal Gallardo-Alba, Bjorn Grüning, Byung June Ko, Alex Ostrovsky, Marc Palmada-Flores, Brandon D. Pickett, Keon Rabbani, Agostinho Antunes, Jennifer R. Balacco, Mark J. P. Chaisson, Haoyu C... | <p>Improvements in genome sequencing and assembly are enabling high-quality reference genomes for all species. However, the assembly process is still laborious, computationally and technically demanding, lacks standards for reproducibility, and is... | | Bioinformatics, ERGA | Alba Marino | 2025-05-06 22:47:39 | ||
22 May 2025

POSTPRINT
The genome sequence of the Violet Carpenter Bee, Xylocopa violacea (Linnaeus, 1785): a hymenopteran species undergoing range expansionA high-quality genome assembly for carpenter beesRecommended by Christian Lopezguerra and Gavin Douglas
Christian Lopezguerra (1) and Gavin M. Douglas (2, 3) (1) Department of Plant and Microbial Biology; (2) Department of Biological Sciences; (3) Bioinformatics Research Center, North Carolina State University, USA Climate change and anthropogenic stressors are driving rapid biodiversity loss and dynamic shifts in species ranges (Outhwaite et al. 2022). Partially in response to the decline in biodiversity, the European Reference Genome Atlas (ERGA) has been generating high-quality accessible genome resources, allowing for a more collaborative network and assisting conservation efforts. One recent target was the violet carpenter bee (Xylocopa violacea; Figure 1), one of the many insects that have shown a recent expansion in their range within Europe. This species is a key pollinator and is therefore of great interest for ecological and agricultural purposes. In addition, anticancer research with melittin variants present in the venom of the violet carpenter bee shows potential (Erkoc et al. 2022; von Reumont et al. 2022). However, genetic analyses have been limited by the prior contig-level assembly of the genome (Koludarov et al. 2023). Developing a high-quality, annotated reference genome for the carpenter bee was the goal of Nash and colleagues’ (2024) research, as part of the European Reference Genome Atlas initiative.
Figure 1: Violet carpenter bee (in Margarida, Spain). Copyright Susanne Vogel, a photographer who made this available on iNaturalist.com. Distributed under a CC-BY 4.0 license. The authors coupled long and short-read sequencing techniques to create an improved assembly. In particular, they used both short-read RNA-seq and long-read Iso-Seq for gene annotation. They also used Hi-C sequencing to capture chromosome conformational information to aid scaffolding. Their final assembly contains 1,300 scaffolds and has a BUSCO completeness level of 99.75% (Manni et al. 2021), aligning with the standards of the European Reference Genome Atlas. The authors generated a 1.02 gigabase assembly, which was much larger than the expected size of 672 megabases based on k-mer content. The authors partially explain the difference by the high repeat content in the genome, particularly specific 109-mer and 217-mer repeats. Due to this high repeat content, the authors could not assemble full chromosomes but instead produced 17 pseudo-chromosomes comprised of 481.4 megabases (in addition to all other unlocalized scaffolds). This high-quality reference genome will be valuable for future studies on population and functional genomics of carpenter bees (Xylocopa). Indeed, this is the first high-quality annotated pseudo-chromosomal genome assembly of the genus Xylocopa, which includes hundreds of other species. It will enable improved investigation into genomic signatures associated with shifting populations. More generally, this reference genome will be useful for comparative analyses with other Hymenoptera species.
References Erkoc P, von Reumont BM, Lüddecke T, Henke M, Ulshöfer T, Vilcinskas A, Fürst R, Schiffmann S (2022) The pharmacological potential of novel melittin variants from the honeybee and solitary bees against inflammation and cancer. Toxins, 14, 818. https://doi.org/10.3390/toxins14120818 Koludarov I, Velasque M, Senoner T, Timm T, Greve C, Hamadou AB, Gupta DK, Lochnit G, Heinzinger M, Vilcinskas A, Gloag R, Harpur BA, Podsiadlowski L, Rost B, Jackson TNW, Dutertre S, Stolle E, von Reumont BM (2023) Prevalent bee venom genes evolved before the aculeate stinger and eusociality. BMC Biology, 21, 229. https://doi.org/10.1186/s12915-023-01656-5 Manni M, Berkeley MR, Seppey M, Simão FA, Zdobnov EM (2021) BUSCO update: Novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Molecular Biology and Evolution, 38, 4647–4654. https://doi.org/10.1093/molbev/msab199 Nash WJ, Man A, McTaggart S, Baker K, Barker T, Catchpole L, Durrant A, Gharbi K, Irish N, Kaithakottil G, Ku D, Providence A, Shaw F, Swarbreck D, Watkins C, McCartney AM, Formenti G, Mouton A, Vella N, von Reumont BM, Vella A, Haerty W (2024) The genome sequence of the Violet Carpenter Bee, Xylocopa violacea (Linnaeus, 1785): a hymenopteran species undergoing range expansion. Heredity, 133, 381–387. https://doi.org/10.1038/s41437-024-00720-2 Outhwaite CL, McCann P, Newbold T (2022) Agriculture and climate change are reshaping insect biodiversity worldwide. Nature, 605, 97–102. https://doi.org/10.1038/s41586-022-04644-x von Reumont BM, Dutertre S, Koludarov I (2022) Venom profile of the European carpenter bee Xylocopa violacea: Evolutionary and applied considerations on its toxin components. Toxicon: X, 14, 100117. https://doi.org/10.1016/j.toxcx.2022.100117
| The genome sequence of the Violet Carpenter Bee, *Xylocopa violacea* (Linnaeus, 1785): a hymenopteran species undergoing range expansion | Will J. Nash, Angela Man, Seanna McTaggart, Kendall Baker, Tom Barker, Leah Catchpole, Alex Durrant, Karim Gharbi, Naomi Irish, Gemy Kaithakottil, Debby Ku, Aaliyah Providence, Felix Shaw, David Swarbreck, Chris Watkins, Ann M. McCartney, Giulio F... | <p style="text-align: justify;">We present a reference genome assembly from an individual male Violet Carpenter Bee (<em>Xylocopa violacea</em>, Linnaeus 1758). The assembly is 1.02 gigabases in span. 48% of the assembly is scaffolded into 17 pseu... | | Arthropods, ERGA, ERGA Pilot | Christian Lopezguerra | 2025-05-16 23:21:44 | ||
21 May 2025
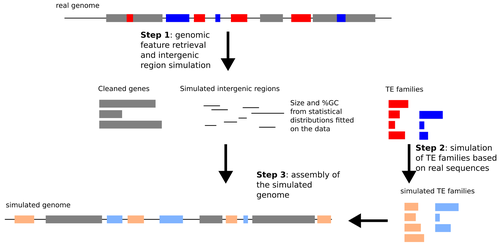
Particular sequence characteristics induce bias in the detection of polymorphic transposable element insertionsA new simulation pipeline enhances benchmarking of transposon polymorphism detection toolsRecommended by Raúl Castanera based on reviews by Tianxiong Yu and 1 anonymous reviewerTransposable Elements (TEs) are one of the main sources of genome variability. However, their study in populations has been hampered by the difficulty of properly detecting them using whole-genome re-sequencing data. Despite the expectations generated by the rise of long-read sequencing, today it is becoming clear that such technologies will not replace short-reads for analyzing large populations in the short term. Detecting Transposon Insertion Polymorphisms (TIPs) from short-read data is a challenging task, due to the repetitive nature of TE sequences that complicate read mapping. Nevertheless, accurate TIP detection is essential for understanding the evolutionary dynamics of TEs, their regulatory roles and their link with phenotypic variability. In the past 15 years, more than 20 tools have been developed for TIP detection using short-read data, but only a few independent benchmarks have been performed so far (Chen et al. 2023; Nelson et al. 2017; Rishishwar et al. 2017; Vendrell-Mir et al. 2019). Previous benchmarks have used simulated and real data to evaluate tool performance, each with its own set of advantages and limitations. In particular, introducing artificial insertions and simulating genomic short-reads may not reflect the nature of real TEs. By contrast, using real TE insertions as benchmarks can introduce bias since TE annotations are never perfect. Verneret et al. (2025) introduce an original, alternative approach in which a comprehensive simulation method mimics the most important sequence features of real TEs and non-TE intergenic regions. This simulated data is then combined with true genic sequences, generating a pseudochromosome that can be used for benchmarking TIP detection pipelines. Using this approach, the authors eliminate the bias of TE annotation on real genomes, while preserving most of the characteristics of natural TEs. Using simulated pseudochromosomes for Drosophila melanogaster and Arabidopsis thaliana, Verneret et al. (2025) found that the performance of 14 commonly used TIP-calling tools is highly variable, with only a few performing well, and only at high sequencing depths. In addition to this, the authors analyzed the sequence features of true-positive and false-positive TIP calls, and found that specific TE sequence characteristics (e.g., length, age, etc.) affect the detection of both reference and non-reference TIPs. The approach described by Verneret et al. (2025) is an important contribution to the field for several reasons. On the one hand, the results shown in the publication will help the users of such tools make informed decisions before launching their experiments. For more advanced users, it will enable future benchmarks to identify which tools perform best for different species, each with their own sequence characteristics. For software developers, the data released constitutes a precious dataset to test their tools in the same conditions. Finally, the identification of sequence characteristics enriched among false positives and false negatives also gives an opportunity for developers to improve the performance of the new tools by considering these specificities.
References Chen J, Basting PJ, Han S, Garfinkel DJ, Bergman CM (2023) Reproducible evaluation of transposable element detectors with McClintock 2 guides accurate inference of Ty insertion patterns in yeast. Mobile DNA, 14, 8. https://doi.org/10.1186/s13100-023-00296-4 Nelson MG, Linheiro RS, Bergman CM (2017) McClintock: An integrated pipeline for detecting transposable element insertions in whole-genome shotgun sequencing data. G3: Genes, Genomes, Genetics, 7, 2763–2778. https://doi.org/10.1534/g3.117.043893 Rishishwar L, Mariño-Ramírez L, Jordan IK (2017) Benchmarking computational tools for polymorphic transposable element detection. Briefings in Bioinformatics, 18, 908–918. https://doi.org/10.1093/bib/bbw072 Vendrell-Mir P, Barteri F, Merenciano M, González J, Casacuberta JM, Castanera R (2019) A benchmark of transposon insertion detection tools using real data. Mobile DNA, 10, 53. https://doi.org/10.1186/s13100-019-0197-9 Verneret M, Le VA, Faraut T, Turpin J, Lerat E (2025) Particular sequence characteristics induce bias in the detection of polymorphic transposable element insertions. bioRxiv, ver. 4 peer-reviewed and recommended by PCI Genomics https://doi.org/10.1101/2024.09.25.614865
| Particular sequence characteristics induce bias in the detection of polymorphic transposable element insertions | Marie Verneret, Van Anthony Le, Thomas Faraut, Jocelyn Turpin, Emmanuelle Lerat | <p>Transposable elements (TEs) have an important role in genome evolution but are challenging for bioinformatics detection due to their repetitive nature and ability to move and replicate within genomes. New sequencing technologies now enable the ... | | Bioinformatics, Evolutionary genomics, Population genomics, Viruses and transposable elements | Raúl Castanera | 2024-09-30 08:29:19 | ||
20 May 2025

Draft genome and transcriptomic sequence data of three invasive insect speciesThree more reference genomes of invasive insect speciesRecommended by Vincent Lacroix based on reviews by Jean-Marc Aury and Nicolas ParisotThe number and prevalence of invasive species have risen in the last decades together with international trade (Hulme 2021). As invasive species, insects have received less attention than plants. In particular, there are fewer reference genomes currently available, although genomic resources can be useful to investigate invasion dynamics and develop more effective management strategies. Lombaeart et al. (2025) sequenced and assembled the genomes of three species: Cydalima perspectalis (the box tree moth), Leptoglossus occidentalis (the western conifer seed bug), and Tecia solanivora (the Guatemalan tuber moth), which have in common their rapid spread and severe impact on their respective host plants (boxwoods, conifers and potatoes). The authors generated PacBio HiFi reads, together with Hi-C data to obtain assemblies meeting international quality standards. They also generated short-read RNA-seq data, which they used to provide initial structural annotations of the genes. The resulting reference genomes are still in a draft state because repeats and heterozygosity are notoriously hard to handle. The most challenging genome to assemble was L. occidentalis, with an estimated size of 1.5 Gb, an estimated repeat content of 58%, and an estimated heterozygosity of 1.8%. The raw data produced can still be analysed more in depth to characterise further the repeat content and the heterozygosity of these species. These reference genomes can readily be used for identifying genetic markers of interest for a variety of applications. In a general context where there is a growing awareness that data production is associated with a significant part of the carbon footprint of research (De Paepe et al. 2024), this dataset has high chances to be extensively reused and analysed by the community.
References De Paepe M, Jeanneau L, Mariette J, Aumont O, Estevez-Torres A (2024) Purchases dominate the carbon footprint of research laboratories. PLOS Sustainability and Transformation, 3, e0000116. https://doi.org/10.1371/journal.pstr.0000116 Hulme, P E (2021) Unwelcome exchange: international trade as a direct and indirect driver of biological invasions worldwide. One Earth 4, 666–679. https://doi.org/10.1016/j.oneear.2021.04.015 Lombaert E, Klopp C, Blin A, Annonay G, Iampietro C, Lluch J, Sallaberry M, Valière S, Poloni R, Joron M, Deleury E (2025) Draft genome and transcriptomic sequence data of three invasive insect species. bioRxiv, ver. 2 peer-reviewed and recommended by PCI Genomics. https://doi.org/10.1101/2024.12.02.626401 | Draft genome and transcriptomic sequence data of three invasive insect species | Eric Lombaert, Christophe Klopp, Aurélie Blin, Gwenolah Annonay, Carole Iampietro, Jérôme Lluch, Marine Sallaberry, Sophie Valière, Riccardo Poloni, Mathieu Joron, Emeline Deleury | <p><em>Cydalima perspectalis</em> (the box tree moth), <em>Leptoglossus occidentalis</em> (the western conifer seed bug), and <em>Tecia solanivora</em> (the Guatemalan tuber moth) are three economically harmful invasive insect species. This study ... | | Arthropods | Vincent Lacroix | Jean-Marc Aury, Nicolas Parisot | 2024-12-06 11:07:34 | |
14 May 2025
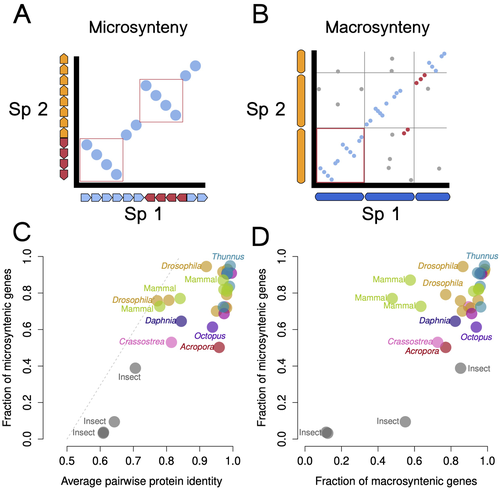
Genomic changes are varied across congeneric species pairs of animalsExploring the correlation between speciation and genome rearrangementsRecommended by Javier del Campo based on reviews by Jean-Baptiste Ledoux and 3 anonymous reviewers
Francis et al. (2025) investigate the relationship between genomic rearrangement, specifically macro- and micro-synteny, and speciation across a broad range of animal phyla. Using chromosome-level genome assemblies, they generated 1:1 ortholog pairs and analyzed synteny conservation using custom bioinformatics pipelines to quantify microsynteny. The study is well written, methodologically sound, and offers valuable insights beyond comparative genomics. The authors show that while most congeneric species pairs exhibit disruptions in micro-synteny, they retain high levels of protein sequence identity. They also find that macro- and micro-synteny decay with speciation but are often decoupled, indicating no universal genomic trajectory during divergence. Their conclusion, that synteny patterns alone are insufficient to define species boundaries (Steenwyk and King 2024), is well supported by their data. The discussion effectively situates the work within the broader context of speciation research. It thoughtfully addresses study limitations, such as challenges in synteny block quantification, chromosomal rearrangement rates, and the scarcity of high-quality genome assemblies. The manuscript also outlines clear directions for future research, including the need for more accurate divergence time estimates and expanded taxonomic sampling (Formenti et al. 2022). Formenti G, Theissinger K, Fernandes C, Bista I, Bombarely A, Bleidorn C, et al. (2022) The era of reference genomes in conservation genomics. Trends in Ecology & Evolution, 37, 197–202. https://doi.org/10.1016/j.tree.2021.11.008 Francis WR, Vargas S, Wörheide G (2025) Genomic changes are varied across congeneric species pairs of animals. bioRxiv, ver. 4 peer-reviewed and recommended by PCI Genomics https://doi.org/10.1101/2024.09.05.611358 Steenwyk JL, King N (2024) The promise and pitfalls of synteny in phylogenomics. PLOS Biology, 22, e3002632. https://doi.org/10.1371/journal.pbio.3002632
| Genomic changes are varied across congeneric species pairs of animals | Warren R. Francis, Sergio Vargas, Gert Wörheide | <p>Synteny, the shared arrangement of genes on chromosomes between related species, is a marker of shared ancestry, and synteny-breaking events can result in genomic incompatibilities between populations and ultimately lead to speciation events. D... | | Evolutionary genomics | Javier del Campo | Anonymous, Nicolas Shogo Locatelli, Jean-Baptiste Ledoux, Anonymous | 2024-09-06 17:57:07 | |
06 May 2025

Comparison of whole-genome assemblies of European river lamprey (Lampetra fluviatilis) and brook lamprey (Lampetra planeri)Phased genomes suggest that L. fluviatilis and L. planeri are two ecotypes of the same speciesRecommended by Samuel Abalde based on reviews by Ricardo C. Rodríguez de la Vega, Quentin Rougemont and 1 anonymous reviewer
Lampreys are the focus of intense research. Together with hagfishes, they form the Cyclostomata, the sister group of jawed vertebrates, and hence they are a key group for disentangling the early evolution of many vertebrate features (Shimel and Donoghue 2012; McCauley et al. 2015). Ecologically, lamprey species show a diverse array of life modes, including parasitic and non-feeding species, and inhabit freshwater and marine habitats or both (i.e. anadromous species; Docker and Potter 2019). One of these anadromous species, the sea lamprey (Petromyzon marinus), took advantage of man-made canals to invade the North American Great Lakes in the early 20th century, decimating many fish populations. Today, the control of these invasive populations is paramount for the survival of the region’s fishing industry (Ferreira-Martins et al. 2021). All these research avenues will benefit from the generation of new genomic data, an invaluable resource in evolutionary and conservation biology. In this manuscript, Tørresen et al. (2025) present phased, chromosome-level assemblies from two lamprey species: the European river lamprey (Lampetra fluviatilis) and the brook lamprey (Lampetra planeri). These two genome assemblies are of high quality and will undoubtedly become a key resource in lamprey research. In particular, the authors showcase the potential of such genomes from two perspectives. First, comparing their assemblies to the already published genomes from P. marinus and another specimen of L. fluviatilis, they propose that lamprey genomes are highly conserved and display large syntenic blocks shared among species. Second, phylogenetic analyses and the annotation of SNPs suggest that L. fluviatilis and L. planeri should be considered two ecotypes of the same species complex, instead of two separate species. This might not be new for anyone knowledgeable in lamprey biology (Rougemont et al. 2017), but it is surprising given the distinct ecology of the two lampreys: L. fluviatilis is a parasitic, anadromous species, whereas L. planeri is a non-feeding, freshwater species. In addition to the biological significance of this manuscript, I would like to acknowledge the robustness of the analytical approaches. These genomes were assembled and annotated following two pipelines recently developed at EBP-Nor, the Norwegian initiative of the Earth BioGenome Project (EBP). These pipelines are designed to be an easy-to-use, end-to-end solution for genomic analyses and are likely to become a standard for the EBP and European Reference Genome Atlas initiatives. There can be no better evidence of their effectiveness than these two phased, chromosome-level, highly complete genome assemblies.
References Docker MF, Potter IC (2019) Life history evolution in lampreys: Alternative migratory and feeding types. In: Docker M (ed) Lampreys: Biology, Conservation and Control. Fish & Fisheries Series, vol 38. Springer, Dordrecht. https://doi.org/10.1007/978-94-024-1684-8_4 Ferreira-Martins D, Champer J, McCauley DW, Zhang Z, Docker MF (2021) Genetic control of invasive sea lamprey in the Great Lakes. Journal of Great Lakes Research, 47, S764-S775. https://doi.org/10.1016/j.jglr.2021.10.018 McCauley DW, Docker MF, Whyard S, Li W (2015) Lampreys as diverse model organisms in the genomics era. BioScience, 65(11), 1046-1056. https://doi.org/10.1093/biosci/biv139 Rougemont Q, Gagnaire PA, Perrier C, Genthon C, Besnard AL, Launey S, Evanno G (2017) Inferring the demographic history underlying parallel genomic divergence among pairs of parasitic and nonparasitic lamprey ecotypes. Molecular Ecology, 26(1), 142-162. https://doi.org/10.1111/mec.13664 Shimeld SM, Donoghue PC (2012) Evolutionary crossroads in developmental biology: cyclostomes (lamprey and hagfish). Development, 139(12), 2091-2099. https://doi.org/10.1242/dev.074716 Tørresen OK, Garmann-Aarhus B, Hoff SNK, Jentoft S, Svensson M, Schartum E, Tooming-Klunderud A, Skage M, Krabberød A, Vøllestad LA, Jakobsen KS (2025) Comparison of whole-genome assemblies of European river lamprey (Lampetra fluviatilis) and brook lamprey (Lampetra planeri). bioRxiv, ver. 5 peer-reviewed and recommended by PCI Genomics https://doi.org/10.1101/2024.12.06.627158 | Comparison of whole-genome assemblies of European river lamprey (*Lampetra fluviatilis*) and brook lamprey (*Lampetra planeri*) | Ole K. Tørresen, Benedicte Garmann-Aarhus, Siv Nam Khang Hoff, Sissel Jentoft, Mikael Svensson, Eivind Schartum, Ave Tooming-Klunderud, Morten Skage, Anders Krabberød, Leif Asbjørn Vøllestad, Kjetill S. Jakobsen | <p>We present haplotype-resolved whole-genome assemblies from one individual European river lamprey (Lampetra fluviatilis) and one individual brook lamprey (Lampetra planeri), usually regarded as sister species. The genome assembly of L. fluviatil... | | Bioinformatics, Evolutionary genomics, Vertebrates | Samuel Abalde | 2024-12-14 14:35:51 | ||
30 Apr 2025
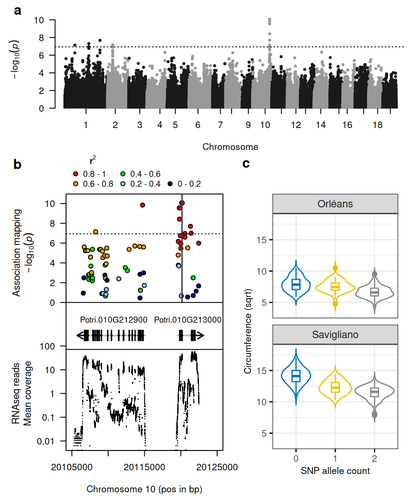
Natural variation in chalcone isomerase defines a major locus controlling radial stem growth variation among Populus nigra populationsAdvancing our understanding of poplar growth using a multi-omics approachRecommended by Wirulda Pootakham based on reviews by Gancho Slavov and 1 anonymous reviewerPoplar is a promising resource, valued not only for wood production and the development of lignocellulosic biomass, but also for its potential role in carbon sequestration. Recognizing the importance of stem growth for wood production and biomass development, Duruflé et al. (2025) present a comprehensive study on the genetic basis of radial stem growth variation in natural populations of black poplar (Populus nigra). They employed a systems biology approach to identify the quantitative trait loci (QTLs) underlying this trait, integrating genomic, transcriptomic, and phenotypic data from a large collection of poplar genotypes. Their genome-wide association study (GWAS) analysis identified single nucleotide polymorphisms linked to two gene models predicted to encode chalcone isomerase, an enzyme involved in the flavonoid pathway. The authors then used the RNA-seq data to test whether the expression of the candidate genes correlated with the phenotypes, and indeed the level of expression of both genes displayed a correlation to the stem circumference. To support their findings, the authors compared the location of the QTLs detected in this study with previously published QTLs. Interestingly, they found a previously reported QTL co-localizing with the newly identified one. The authors have addressed the concerns raised by reviewers on the GWAS analysis and discussed the complication of this QTL study in the manuscript. In essence, the authors have combined the power of GWAS and transcriptomics to locate candidate genes and applied population genetics to explore the evolutionary context of the identified gene. This comprehensive approach provides strong evidence for the role of chalcone isomerase in controlling radial stem growth variation in black poplar. The study opens up avenues for further research into the precise mechanisms by which chalcone isomerase and flavonoid metabolism influence stem growth and provides useful information for future poplar breeding programs.
References Duruflé H, Déjardin A, Jorge V, Pégard M, Pilate G, Rogier O, Sanchez L, Segura V (2025) Natural variation in chalcone isomerase defines a major locus controlling radial stem growth variation among Populus nigra populations. bioRxiv, ver. 3 peer-reviewed and recommended by PCI Genomics. https://doi.org/10.1101/2024.10.21.618920 | Natural variation in chalcone isomerase defines a major locus controlling radial stem growth variation among *Populus nigra* populations | Harold Durufle, Annabelle Dejardin, Veronique Jorge, Marie Pegard, Gilles Pilate, Odile Rogier, Leopoldo Sanchez, Vincent Segura | <p>Poplar is a promising resource for wood production and the development of lignocellulosic biomass, but currently available varieties have not been optimized for these purposes. Therefore, it is critical to investigate the genetic variability an... | | Plants, Population genomics | Wirulda Pootakham | Fernando Guerra , Gancho Slavov | 2024-10-25 09:37:10 | |
13 Mar 2025
Estimating allele frequencies, ancestry proportions and genotype likelihoods in the presence of mapping biasA novel genotype likelihood-based method to reduce mapping bias in low-coverage and ancient DNA studiesRecommended by Sebastian Ernesto Ramos-Onsins based on reviews by Maxime Lefebvre, Michael Westbury and Adrien Oliva
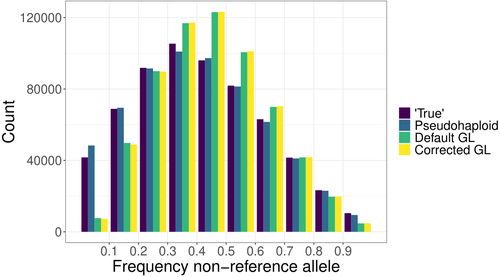
The study of genomic variability within and between populations, as well as among species, relies on comparative analyses of homologous positions—sites that share a common evolutionary origin. Homology is inferred through sequence similarity (Reeck et al. 1987). However, the ability to detect homologous regions can be compromised when sequence mismatches accumulate due to mutations, especially when analyzing short DNA fragments, as in short-read sequencing (Li et al. 2008). In the genomic era, accurately mapping homologous DNA fragments to a reference genome is essential for obtaining precise estimates of genetic variability and evolutionary inferences (e.g., Li et al. 2008; Ellegren 2014). However, short-read, high-throughput sequencing often introduces mapping bias, disproportionately favoring the reference allele. This bias distorts allele frequency estimates, ancestry proportions, and genotype likelihoods, impacting downstream analyses (e.g., Günther & Nettelblad 2019; Martiniano et al. 2020). Mapping bias is particularly problematic in ancient DNA studies, where post-mortem damage exacerbates sequencing errors. DNA fragmentation limits read length, while deamination, causing G to A and C to U transitions, increases mismatches and further complicates homology identification (Dabney & Pääbo 2013). These degradation processes contribute to the misidentification of true variants, confounding evolutionary inferences. Various strategies have been developed to mitigate mapping bias, including the commonly used approach, called pseudo-haploid data, that randomly picks a single read at each analyzed position for each individual, thereby retaining a single allele at each polymorphic site (Günther & Nettelblad 2019; Barlow et al. 2020). Günther et al. (2025) introduce a novel method to correct mapping bias using a genotype likelihood-based approach, incorporating a mapping bias ratio to adjust for reference allele overrepresentation. The method specifically targets known single nucleotide polymorphisms (SNPs) because in population genomic analysis of ancient DNA data, low coverage and post-mortem damage often hinder the ability to identify novel SNPs in most individuals. The analysis focuses on DNA fragmentation, assuming that deamination effects are minimal when considering ascertained SNPs. The proposed method was compared against existing approaches, including pseudo-haploid data and standard genotype likelihood-based probabilistic methods. The evaluation was performed using both empirical and simulated data. For empirical data, low-coverage sequencing data from the 1000 Genomes Project (Finnish in Finland, Japanese in Tokyo, Yoruba in Ibadan, Nigeria populations) was analyzed, while for simulated data, ancient DNA-like datasets were generated using ms-prime (Kelleher et al. 2016), modeling different sequencing depths, divergence times, and reference genome choices. The study assesses the impact of mapping bias on the ratio of reference versus non-reference allele mapping, the accuracy of SNP allele frequency estimates relative to true frequencies, the deviation and variance between estimated and true allele frequencies, population differentiation and the estimation of admixture proportions using supervised and unsupervised methods, considering both genotype likelihoods and genotype calls. Günther et al. (2025) bring to light that all methods analyzed exhibit minor but systematic reference allele bias. The new corrected genotype likelihood method outperforms the standard genotype likelihood approach in correlating with true allele frequencies, although the pseudo-haploid method still provides the most accurate estimates. Mapping bias also affects ancestry estimation, leading to admixture proportion errors of up to 4%, though this effect is smaller than the 10% discrepancy observed across different inference methods. The work performed by Günther et al. (2025) provides a rigorous and innovative evaluation of mapping bias in the context of ascertained SNPs, introducing a probabilistic approach that improves bias correction. Unlike non-probabilistic methods such as pseudo-haploid data, the genotype likelihood framework leverages all sequencing reads for each analyzed SNP, and can incorporate additional bias corrections, enhancing its applicability across different sequencing conditions. While probabilistic approaches offer clear advantages in bias correction, they can be less intuitive to interpret compared to traditional genotype calling methods. This study highlights that mapping bias is pervasive across all methods, influencing evolutionary inferences such as selection signals and population differentiation. Although the improvements in allele frequency recovery may seem modest, the genome-wide impact of mapping bias is significant, especially in ancient DNA studies, making bias correction essential for robust evolutionary analyses.
References Ellegren H. (2014) Genome sequencing and population genomics in non-model organisms. Trends Ecol Evol. 29(1):51-63. https://doi.org/10.1016/j.tree.2013.09.008 Günther T, Nettelblad C. (2019) The presence and impact of reference bias on population genomic studies of prehistoric human populations. PLoS Genet.15(7):e1008302. https://doi.org/10.1371/journal.pgen.1008302 Günther T., Goldberg A., Schraiber J. G. (2025) Estimating allele frequencies, ancestry proportions and genotype likelihoods in the presence of mapping bias. bioRxiv, ver. 5 peer-reviewed and recommended by PCI Genomics https://doi.org/10.1101/2024.07.01.601500 Kelleher J., Etheridge A. M., McVean G. (2016) Efficient coalescent simulation and genealogical analysis for large sample sizes. PLoS computational biology, 12(5):e1004842. https://doi.org/10.1371/journal.pcbi.1004842 Li H, Ruan J, Durbin R. (2008) Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 18(11):1851-8. https://doi.org/10.1101/gr.078212.108 Reeck GR, de Haën C, Teller DC, Doolittle RF, Fitch WM, Dickerson RE, et al. (1987) "Homology" in proteins and nucleic acids: a terminology muddle and a way out of it. Cell. 50 (5): 667. https://doi.org/10.1016/0092-8674(87)90322-9 | Estimating allele frequencies, ancestry proportions and genotype likelihoods in the presence of mapping bias | Torsten Günther, Amy Goldberg, Joshua G. Schraiber | <p>Population genomic analyses rely on an accurate and unbiased characterization of the genetic composition of the studied population. For short-read, high-throughput sequencing data, mapping sequencing reads to a linear reference genome can bias ... | | Bioinformatics, Evolutionary genomics, Population genomics | Sebastian Ernesto Ramos-Onsins | 2024-07-02 10:46:19 | ||
10 Mar 2025

hdmax2, an R package to perform high dimension mediation analysisHigh-dimensional mediation analysis: Unraveling pathways linking external exposures to health outcomesRecommended by Guillaume Laval based on reviews by Pierre Neuvial and Gaspard KernerPittion et al. (2025) introduce an R package called hdmax2, which implements an enhanced version of the “High-Dimensional Mediation Analysis using the Max-Squared” (HDMAX2) method originally proposed by Jumentier et al. (2023) for high-dimensional mediation analysis. The goal of mediation analysis is to quantify the indirect effect of a variable M in the causal relationship between exposure X and outcome Y. The fundamental concept behind HDMAX2 methods is to use a latent factor mixed model to estimate the effects of unobserved confounders and a max-squared test to identify significant mediators. The HDMAX2 method represents a significant advancement in the case of high-dimensional mediation, such as DNA methylation or gene expression analysis, where the number of mediators often far exceeds the sample size. The main contributions of this article are the implementation of the HDMAX2 method as an R package, and an extension of the original method to binary outcomes and to binary, categorical, and multivariate exposures, as opposed to only continuous variables. The package includes visualization tools, helper functions for mediator selection, and options for handling multivariate exposures. A key strength of the package lies in its versatility. The new package, hdmax2, accommodates a variety of data types. This flexibility makes it a valuable tool for researchers analyzing high-throughput molecular data. Finally to illustrate this flexibility, the authors present two case studies that were not described in the Jumentier et al. (2023) analysis. In the first case study, the authors employed mediation analysis to assess the potential causal role of DNA methylation in the pathway linking the HER2 status of breast cancer (a marker for an aggressive breast cancer subtype) to a survival risk score, which was derived from a six-gene expression signature and is inversely correlated with patient survival. In the second case study, the authors conducted mediation analysis to explore the role of gene expression in the pathway linking patient gender to the occurrence of multiple sclerosis specific subtypes: clinically isolated syndrome and relapsing-remitting multiple sclerosis. These illustrate the relevance of hdmax2 to study the transcriptome and the methylome. In conclusion, the hdmax2 R package will be invaluable for handling high-dimensional molecular data in the study of the intricate pathways through which exposures influence health outcomes.
References Jumentier B, Barrot C-C, Estavoyer M, Tost J, Heude B, François O, Lepeule J (2023) High-dimensional mediation analysis: A new method applied to maternal smoking, placental DNA methylation, and birth outcomes. Environmental Health Perspectives, 131, 047011. https://doi.org/10.1289/EHP11559 Pittion F, Jumentier B, Nakamura A, Lepeule J, Francois O, Richard M (2025) hdmax2, an R package to perform high dimension mediation analysis. HAL, ver. 4 peer-reviewed and recommended by PCI Genomics. https://hal.science/hal-04658960 | hdmax2, an R package to perform high dimension mediation analysis | Florence Pittion, Basile Jumentier, Aurélie Nakamura, Johanna Lepeule, Olivier François, Magali Richard | <p>Mediation analysis plays a crucial role in epidemiology, unraveling the intricate pathways through which exposures exert influence on health outcomes. Recent advances in high-throughput sequencing techniques have generated growing interest in a... | | Bioinformatics | Guillaume Laval | 2024-09-10 11:49:02 | ||
26 Feb 2025
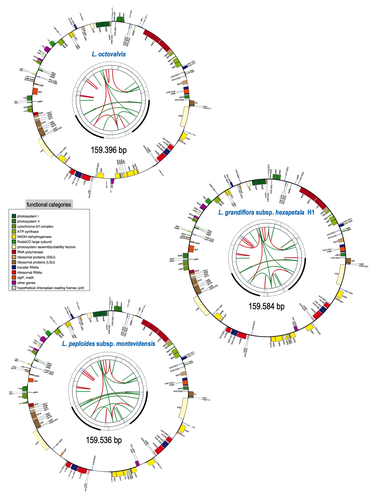
Sequencing, de novo assembly of Ludwigia plastomes, and comparative analysis within the Onagraceae familyOnagre, monster, invasion and geneticsRecommended by Francois Sabot based on reviews by 2 anonymous reviewers
The first time I heard of ”onagres” in French was when I was a teenager, through the books of Pierre Bordage as fantastic monsters, or through historical games as Roman siege weapons (onagers). At this time, I was far from imagining that “onagre” also refers to a very large flowering plant family, as it is the French term for evening primroses. In this family, the genus Ludwigia comprises species that are invasive (resembling in that way the ancient armies using onagers to invade cities) in aquatic environments, degrading ecosystems already fragilized by human activities. To counteract this phenomenon, it is of high importance to understand their propagation of these species. However, the knowledge about their genetics and diversity is very scarce, and thus tracking their dispersal using genetic information is complicated, and in fact almost impossible. Barloy-Hubler et al. (2024) proposed in the present manuscript a new set of chloroplastic genomes from two of these species, Ludwigia grandiflora subsp. hexapetala and Ludwigia peploides subsp. montevidensis, and compared them to the published chloroplastic genome of Ludwigia octovalis. They explored the possibility of assembling these genomes relying solely on short reads and showed that long reads were necessary to obtain an almost complete assembly for these plastid genomes. In addition, through this approach, they detected two haplotypes in Ludwigia grandiflora subsp. hexapetala as compared to one in a short-read assembly. This highlights the need for long reads data to assess the structure and diversity of chloroplastic genomes. The authors were also able to clarify the phylogeny of the genus Ludwigia. Finally, they identified multiple potential single nucleotide polymorphisms and simple sequence repeats for future evaluation of diversity and dispersal of those invasive species. This analysis, while appearing more technical than biological at first glance, is in fact of high importance for the understanding of ecology and preservation of fragile ecosystems, such as the European watersheds. Indeed, new scientific results and insights are generally linked to a reevaluation of previously analyzed data or samples through new technologies, and this paper is a quite clever example of that matter.
References Barloy-Hubler F, Gac A-LL, Boury C, Guichoux E, Barloy D (2024) Sequencing, de novo assembly of Ludwigia plastomes, and comparative analysis within the Onagraceae family. bioRxiv, ver. 5 peer-reviewed and recommended by PCI Genomics. https://doi.org/10.1101/2023.10.20.563230 Bordage, P (1993) Les Guerriers du Silence, L'Atalante, ISBN 9782905158697
| Sequencing, de novo assembly of *Ludwigia* plastomes, and comparative analysis within the Onagraceae family | F Barloy-Hubler, A-L Le Gac, C Boury, E Guichoux, D Barloy | <p>The Onagraceae family, which belongs to the order Myrtales, consists of approximately 657 species and 17 genera. This family includes the genus <em>Ludwigia </em>L., which is comprised of 82 species. In this study, we focused on the two aquatic... | | Bioinformatics, Plants | Francois Sabot | 2023-12-12 18:05:20 |





