
Latest recommendations
| Id | Title * | Authors * | Abstract * | Picture * | Thematic fields * | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
26 Jun 2024
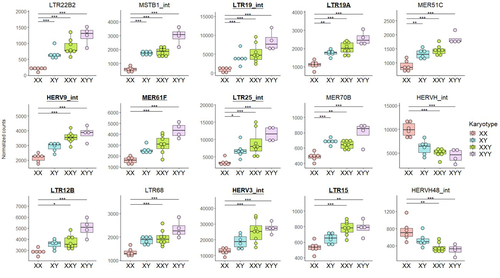
Transposable element expression with variation in sex chromosome number supports a toxic Y effect on human longevityThe number of Y chromosomes is positively associated with transposable element expression in humans, in line with the toxic Y hypothesisRecommended by Anna-Sophie Fiston-Lavier based on reviews by 3 anonymous reviewers based on reviews by 3 anonymous reviewers
The study of human longevity has long been a source of fascination for scientists, particularly in relation to the genetic factors that contribute to differences in lifespan between the sexes. One particularly intriguing area of research concerns the Y chromosome and its impact on male longevity. The Y chromosome expresses genes that are essential for male development and reproduction. However, it may also influence various physiological processes and health outcomes. It is therefore of great importance to investigate the impact of the Y chromosome on longevity. This may assist in elucidating the biological mechanisms underlying sex-specific differences in aging and disease susceptibility. As longevity research progresses, the Y chromosome's role presents a promising avenue for elucidating the complex interplay between genetics and aging. Transposable elements (TEs), often referred to as "jumping genes", are DNA sequences that can move within the genome, potentially causing mutations and genomic instability. In young, healthy cells, various mechanisms, including DNA methylation and histone modifications, suppress TE activity to maintain genomic integrity. However, as individuals age, these regulatory mechanisms may deteriorate, leading to increased TE activity. This dysregulation could contribute to age-related genomic instability, cellular dysfunction, and the onset of diseases such as cancer. Understanding how TE repression changes with age is crucial for uncovering the molecular underpinnings of aging (De Cecco et al. 2013; Van Meter et al. 2014). The lower recombination rates observed on Y chromosomes result in the accumulation of TE insertions, which in turn leads to an enrichment of TEs and potentially higher TE activity. To ascertain whether the number of Y chromosomes is associated with TE activity in humans, Teoli et al. (2024) studied the TE expression level, as a proxy of the TE activity, in several karyotype compositions (i.e. with differing numbers of Y chromosomes). They used transcriptomic data from blood samples collected in 24 individuals (six females 46,XX, six males 46,XY, eight males 47,XXY and four males 47,XYY). Even though they did not observe a significant correlation between the number of Y chromosomes and TE expression, their results suggest an impact of the presence of the Y chromosome on the overall TE expression. The presence of Y chromosomes also affected the type (family) of TE present/expressed. To ensure that the TE expression level was not biased by the expression of a gene in proximity due to intron retention or pervasive intragenic transcription, the authors also tested whether the TE expression variation observed between the different karyotypes could be explained by gene (i.e. here non-TE gene) expression. As TE repression mechanisms are known to decrease over time, the authors also tested whether TE repression is weaker in older individuals, which would support a compelling link between genomic stability and aging. They investigated the TE expression differently between males and females, hypothesizing that old males should exhibit a stronger TE activity than old females. Using selected 45 males (47,XY) and 35 females (46,XX) blood samples of various ages (from 20 to 70) from the Genotype-Tissue Expression (GTEx) project, the authors studied the effect of age on TE expression using 10-year range to group the study subjects. Based on these data, they fail to find an overall increase of TE expression in old males compared to old females. Notwithstanding the small number of samples, the study is well-designed and innovative, and its findings are highly promising. It marks an initial step towards understanding the impact of Y-chromosome ‘toxicity’ on human longevity. Despite the relatively small sample size, which is a consequence of the difficulty of obtaining samples from individuals with sex chromosome aneuploidies, the results are highly intriguing and will be of interest to a broad range of biologists.
References De Cecco M, Criscione SW, Peckham EJ, Hillenmeyer S, Hamm EA, Manivannan J, Peterson AL, Kreiling JA, Neretti N, Sedivy JM (2013) Genomes of replicatively senescent cells undergo global epigenetic changes leading to gene silencing and activation of transposable elements. Aging Cell, 12, 247–256. https://doi.org/10.1111/acel.12047 Teoli J, Merenciano M, Fablet M, Necsulea A, Siqueira-de-Oliveira D, Brandulas-Cammarata A, Labalme A, Lejeune H, Lemaitre J-F, Gueyffier F, Sanlaville D, Bardel C, Vieira C, Marais GAB, Plotton I (2024) Transposable element expression with variation in sex chromosome number supports a toxic Y effect on human longevity. bioRxiv, ver. 5 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2023.08.03.550779 Van Meter M, Kashyap M, Rezazadeh S, Geneva AJ, Morello TD, Seluanov A, Gorbunova V (2014) SIRT6 represses LINE1 retrotransposons by ribosylating KAP1 but this repression fails with stress and age. Nature Communications, 5, 5011. https://doi.org/10.1038/ncomms6011
| Transposable element expression with variation in sex chromosome number supports a toxic Y effect on human longevity | Jordan Teoli, Miriam Merenciano, Marie Fablet, Anamaria Necsulea, Daniel Siqueira-de-Oliveira, Alessandro Brandulas-Cammarata, Audrey Labalme, Hervé Lejeune, Jean-François Lemaitre, François Gueyffier, Damien Sanlaville, Claire Bardel, Cristina Vi... | <p>Why women live longer than men is still an open question in human biology. Sex chromosomes have been proposed to play a role in the observed sex gap in longevity, and the Y male chromosome has been suspected of having a potential toxic genomic ... | | Evolutionary genomics | Anna-Sophie Fiston-Lavier | Anonymous, Igor Rogozin , Paul Jay , Anonymous | 2023-08-18 15:01:38 | |
11 May 2024
The European Reference Genome Atlas: piloting a decentralised approach to equitable biodiversity genomicsInformed Choices, Cohesive Future: Decisions and Recommendations for ERGARecommended by Jitendra Narayan based on reviews by Justin Ideozu and Eric Crandall
The European Reference Genome Atlas (ERGA) (Mc Cartney et al, 2024, Mazzoni et al, 2023) demonstrates the collaborative spirit and intellectual abilities of researchers from 33 European countries. This ambitious project, which is part of the Earth BioGenome Project (Lewin et al., 2018) Phase II, has embarked on an unprecedented mission: to decipher the genetic makeup of 150,000 species over a span of four years. At the heart of ERGA is a decentralized pilot infrastructure specifically built to assist the production of high-quality reference genomes. This structure acts as a scaffold for the massive task of genome sequencing, giving the necessary framework to manage the complexity of genomic research. The research paper under consideration offers a comprehensive narrative of ERGA's evolution, outlining both successes and challenges encountered along the road. One of the most significant issues addressed in the manuscript is the equitable distribution of resources and expertise among participating laboratories and countries. In a project of this magnitude, it is critical to leverage the pooled talents and capacities of researchers from across Europe. ERGA's pan-European network promotes communications and collaboration, creating an environment in which knowledge flows freely and barriers are overcome. This adoption of strong coordination and communication tactics will be essential to ERGA's success. Scientific collaboration depends on efficient communication channels because they allow researchers to share resources, collaborate on new initiatives, and exchange ideas. Through a diverse range of gatherings, courses, and virtual discussion boards, ERGA fosters an environment of transparency and cooperation among members, enabling scientists to overcome challenges and make significant discoveries. The importance ERGA places on training and information transfer programmes is a pillar of its strategy. Understanding the importance of capacity development, ERGA invests in providing researchers with the knowledge and abilities necessary for effectively navigating the complicated terrain of genomic research. A wide range of subjects are covered in training programmes (Larivière et al. 2023), from sample preparation and collection to data processing methods and sequencing technology. Through the development of a group of highly qualified experts, ERGA creates the foundation for continued advancement and creativity in the genomics sector. This manuscript also covers in detail the technological workflows and sequencing techniques used in ERGA's pilot infrastructure. With the aid of cutting-edge sequencing technologies based on both long-read and short-read sequencing, they are working to unravel the complex structure of the genetic code with a level of accuracy and precision never before possible. To guarantee the accuracy of genetic data and prevent mistakes and flaws that can jeopardize the findings' integrity, quality control methods are put in place. Despite having a focus on genome sequencing due to its technological complexities, ERGA also remains firm in its dedication to metadata collection and sample validation. Metadata serves as a critical link between raw genetic data and useful scientific insights, giving necessary context and allowing researchers to draw practical findings from their investigations. Sample validation approaches improve the reliability and reproducibility of the results, providing users confidence in the quality of the genetic data provided by ERGA. Looking ahead, ERGA envisions its decentralized infrastructure serving as a model for global collaborative research efforts. By embracing diversity, encouraging cooperation, and pushing for open access to data and resources, ERGA hopes to catalyze scientific discovery and generate positive change in the field of biodiversity genomics. ERGA aims to promote a more equitable and sustainable future for all by ongoing interaction with stakeholders, intensive outreach and education activities, and policy change advocacy. In addition to its immediate goals, ERGA considers the long-term implications of its work. As genomic technology progresses, the potential application of high-quality reference genomes will continue to grow. From informing conservation efforts and illuminating evolutionary histories to revolutionizing healthcare and agriculture, it is likely that ERGA's contributions will have far-reaching consequences for people and the planet as a whole. Furthermore, ERGA understands the importance of interdisciplinary collaboration in addressing the difficult challenges of the twenty-first century. ERGA aims to integrate genetic research into larger initiatives to promote sustainability and biodiversity conservation by forming relationships with stakeholders from other areas, such as policymakers, conservationists, and indigenous groups. Through shared knowledge and community action, ERGA seeks to create a future in which mankind coexists peacefully with the natural world, guided by a thorough grasp of its genetic legacy and ecological interconnectivity. Finally, the manuscript exemplifies ERGA's collaborative ambitions and achievements, capturing the spirit of creativity and collaboration that defines this ground-breaking effort. As ERGA continues to push the boundaries of genetic research, it remains dedicated to scientific excellence, inclusivity, and the quest of knowledge for the benefit of society. I wholeheartedly recommend the publication of this groundbreaking initiative, offering my enthusiastic endorsement for its valuable contribution to the scientific community. References Lewin, H. A., Robinson, G. E., Kress, W. J., Baker, W. J., Coddington, J., Crandall, K. A., Durbin, R., Edwards, S. V., Forest, F., Gilbert, M. T. P., Goldstein, M. M., Grigoriev, I. V., Hackett, K. J., Haussler, D., Jarvis, E. D., Johnson, W. E., Patrinos, A., Richards, S., Castilla-Rubio, J. C., … Zhang, G. (2018). Earth BioGenome Project: Sequencing life for the future of life. Proceedings of the National Academy of Sciences, 115(17), 4325–4333. https://doi.org/10.1073/pnas.1720115115 Mazzoni, C. J., Claudio, C.i, Waterhouse, R. M. (2023). Biodiversity: an atlas of European reference genomes. Nature 619 : 252-252. https://doi.org/10.1038/d41586-023-02229-w Mc Cartney, A. M., Formenti, G., Mouton, A., Panis, D. de, Marins, L. S., Leitão, H. G., Diedericks, G., Kirangwa, J., Morselli, M., Salces-Ortiz, J., Escudero, N., Iannucci, A., Natali, C., Svardal, H., Fernández, R., Pooter, T. de, Joris, G., Strazisar, M., Wood, J., … Mazzoni, C. J. (2024). The European Reference Genome Atlas: piloting a decentralised approach to equitable biodiversity genomics. bioRxiv, ver. 4 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2023.09.25.559365 | The European Reference Genome Atlas: piloting a decentralised approach to equitable biodiversity genomics | Ann M Mc Cartney, Giulio Formenti, Alice Mouton, Claudio Ciofi, Robert M Waterhouse, Camila J Mazzoni, Diego De Panis, Luisa S Schlude Marins, Henrique G Leitao, Genevieve Diedericks, Joseph Kirangwa, Marco Morselli, Judit Salces, Nuria Escudero, ... | <p>English: A global genome database of all of Earth's species diversity could be a treasure trove of scientific discoveries. However, regardless of the major advances in genome sequencing technologies, only a tiny fraction of species have genomic... | | Bioinformatics, ERGA Pilot | Jitendra Narayan | Justin Ideozu, Eric Crandall | 2023-10-01 01:03:58 | |
01 May 2024
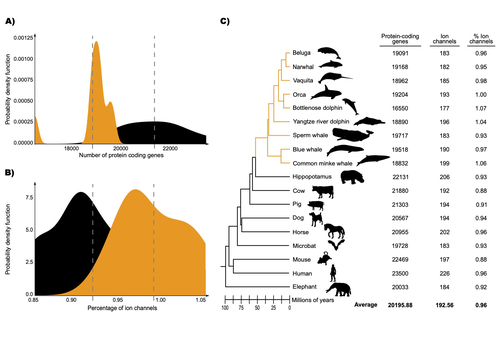
Evolution of ion channels in cetaceans: A natural experiment in the tree of lifePositive selection acted upon cetacean ion channels during the aquatic transitionRecommended by Gavin Douglas based on reviews by 2 anonymous reviewers
The transition of cetaceans (whales, dolphins, and porpoises) from terrestrial to aquatic lifestyles is a striking example of natural selection driving major phenotypic changes (Figure 1). For instance, cetaceans have evolved the ability to withstand high pressure and to store oxygen for long periods, among other adaptations (Das et al. 2023). Many phenotypic changes, such as shifts in organ structure, have been well-characterized through fossils (Thewissen et al. 2009). Although such phenotypic transitions are now well understood, we have only a partial understanding of the underlying genetic mechanisms. Scanning for signatures of adaptation in genes related to phenotypes of interest is one approach to better understand these mechanisms. This was the focus of Uribe and colleagues’ (2024) work, who tested for such signatures across cetacean protein-coding genes.
Figure 1: The skeletons of Ambulocetus (an early whale; top) and Pakicetus (the earliest known cetacean, which lived about 50 million years ago; bottom). Copyright: J. G. M. Thewissen. Displayed here with permission from the copyright holder.
The authors were specifically interested in investigating the evolution of ion channels, as these proteins play fundamental roles in physiological processes. An important aspect of their work was to develop a bioinformatic pipeline to identify orthologous ion channel genes across a set of genomes. After applying their bioinformatic workflow to 18 mammalian species (including nine cetaceans), they conducted tests to find out whether these genes showed signatures of positive selection in the cetacean lineage. For many ion channel genes, elevated ratios of non-synonymous to synonymous substitution rates were detected (for at least a subset of sites, and not necessarily the entire coding region of the genes). The genes concerned were enriched for several functions, including heart and nervous system-related phenotypes. One top gene hit among the putatively selected genes was SCN5A, which encodes a sodium channel expressed in the heart. Interestingly, the authors noted a specific amino acid replacement, which is associated with sensitivity to the toxin tetrodotoxin in other lineages. This substitution appears to have occurred in the common ancestor of toothed whales, and then was reversed in the ancestor of bottlenose dolphins. The authors describe known bottlenose dolphin interactions with toxin-producing pufferfish that could result in high tetrodotoxin exposure, and thus perhaps higher selection for tetrodotoxin resistance. Although this observation is intriguing, the authors emphasize it requires experimental confirmation. The authors also recapitulated the previously described observation (Yim et al. 2014; Huelsmann et al. 2019) that cetaceans have fewer protein-coding genes compared to terrestrial mammals, on average. This signal has previously been hypothesized to partially reflect adaptive gene loss. For example, specific gene loss events likely decreased the risk of developing blood clots while diving (Huelsmann et al. 2019). Uribe and colleagues also considered overall gene turnover rate, which encompasses gene copy number variation across lineages, and found the cetacean gene turnover rate to be three times higher than that of terrestrial mammals. Finally, they found that cetaceans have a higher proportion of ion channel genes (relative to all protein-coding genes in a genome) compared to terrestrial mammals. Similar investigations of the relative non-synonymous to synonymous substitution rates across cetacean and terrestrial mammal orthologs have been conducted previously, but these have primarily focused on dolphins as the sole cetacean representative (McGowen et al. 2012; Nery et al. 2013; Sun et al. 2013). These projects have also been conducted across a large proportion of orthologous genes, rather than a subset with a particular function. Performing proteome-wide investigations can be valuable in that they summarize the genome-wide signal, but can suffer from a high multiple testing burden. More generally, investigating a more targeted question, such as the extent of positive selection acting on ion channels in this case, or on genes potentially linked to cetaceans’ increased brain sizes (McGowen et al. 2011) or hypoxia tolerance (Tian et al. 2016), can be easier to interpret, as opposed to summarizing broader signals. However, these smaller-scale studies can also experience a high multiple testing burden, especially as similar tests are conducted across numerous studies, which often is not accounted for (Ioannidis 2005). In addition, integrating signals across the entire genome will ultimately be needed given that many genetic changes undoubtedly underlie cetaceans’ phenotypic diversification. As highlighted by the fact that past genome-wide analyses have produced some differing biological interpretations (McGowen et al. 2012; Nery et al. 2013; Sun et al. 2013), this is not a trivial undertaking. Nonetheless, the work performed in this preprint, and in related research, is valuable for (at least) three reasons. First, although it is a challenging task, a better understanding of the genetic basis of cetacean phenotypes could have benefits for many aspects of cetacean biology, including conservation efforts. In addition, the remarkable phenotypic shifts in cetaceans make the question of what genetic mechanisms underlie these changes intrinsically interesting to a wide audience. Last, since the cetacean fossil record is especially well-documented (Thewissen et al. 2009), cetaceans represent an appealing system to validate and further develop statistical methods for inferring adaptation from genetic data. Uribe and colleagues’ (2024) analyses provide useful insights relevant to each of these points, and have generated intriguing hypotheses for further investigation.
Das K, Sköld H, Lorenz A, Parmentier E (2023) Who are the marine mammals? In: “Marine Mammals: A Deep Dive into the World of Science”. Brennecke D, Knickmeier K, Pawliczka I, Siebert U, Wahlberg, M (editors). Springer, Cham. p. 1–14. https://doi.org/10.1007/978-3-031-06836-2_1 Huelsmann M, Hecker N, Springer MS, Gatesy J, Sharma V, Hiller M (2019) Genes lost during the transition from land to water in cetaceans highlight genomic changes associated with aquatic adaptations. Science Advances, 5, eaaw6671. https://doi.org/10.1126/sciadv.aaw6671 Ioannidis JPA (2005) Why most published research findings are false. PLOS Medicine, 2, e124. https://doi.org/10.1371/journal.pmed.0020124 McGowen MR, Montgomery SH, Clark C, Gatesy J (2011) Phylogeny and adaptive evolution of the brain-development gene microcephalin (MCPH1) in cetaceans. BMC Evolutionary Biology, 11, 98. https://doi.org/10.1186/1471-2148-11-98 McGowen MR, Grossman LI, Wildman DE (2012) Dolphin genome provides evidence for adaptive evolution of nervous system genes and a molecular rate slowdown. Proceedings of the Royal Society B: Biological Sciences, 279, 3643–3651. https://doi.org/10.1098/rspb.2012.0869 Nery MF, González DJ, Opazo JC (2013) How to make a dolphin: molecular signature of positive selection in cetacean genome. PLOS ONE, 8, e65491. https://doi.org/10.1371/journal.pone.0065491 Sun YB, Zhou WP, Liu HQ, Irwin DM, Shen YY, Zhang YP (2013) Genome-wide scans for candidate genes involved in the aquatic adaptation of dolphins. Genome Biology and Evolution, 5, 130–139. https://doi.org/10.1093/gbe/evs123 Tian R, Wang Z, Niu X, Zhou K, Xu S, Yang G (2016) Evolutionary genetics of hypoxia tolerance in cetaceans during diving. Genome Biology and Evolution, 8, 827–839. https://doi.org/10.1093/gbe/evw037 Thewissen JGM, Cooper LN, George JC, Bajpai (2009) From land to water: the origin of whales, dolphins, and porpoises. Evolution: Education and Outreach, 2, 272–288. https://doi.org/10.1007/s12052-009-0135-2 Uribe C, Nery M, Zavala K, Mardones G, Riadi G, Opazo J (2024) Evolution of ion channels in cetaceans: A natural experiment in the tree of life. bioRxiv, ver. 8 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2023.06.15.545160 Yim HS, Cho YS, Guang X, Kang SG, Jeong JY, Cha SS, Oh HM, Lee JH, Yang EC, Kwon KK, et al. (2014) Minke whale genome and aquatic adaptation in cetaceans. Nature Genetics, 46, 88–92. https://doi.org/10.1038/ng.2835
| Evolution of ion channels in cetaceans: A natural experiment in the tree of life | Cristóbal Uribe, Mariana F Nery, Kattina Zavala, Gonzalo A Mardones, Gonzalo Riadi, Juan C. Opazo | <p>Cetaceans could be seen as a natural experiment within the tree of life in which a mammalian lineage changed from terrestrial to aquatic habitats. This shift involved extensive phenotypic modifications, which represent an opportunity to explore... | | Evolutionary genomics | Gavin Douglas | 2023-07-04 20:53:46 | ||
15 Mar 2024
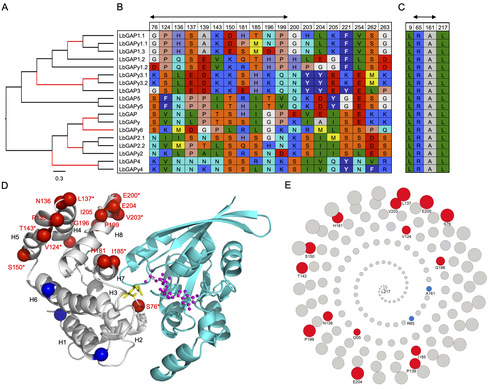
Convergent origin and accelerated evolution of vesicle-associated RhoGAP proteins in two unrelated parasitoid waspsUsing transcriptomics and proteomics to understand the expansion of a secreted poisonous armoury in parasitoid wasps genomesRecommended by Ignacio Bravo based on reviews by Inacio Azevedo and 2 anonymous reviewers
Parasitoid wasps lay their eggs inside another arthropod, whose body is physically consumed by the parasitoid larvae. Phylogenetic inference suggests that Parasitoida are monophyletic, and that this clade underwent a strong radiation shortly after branching off from the Apocrita stem, some 236 million years ago (Peters et al. 2017). The increase in taxonomic diversity during evolutionary radiations is usually concurrent with an increase in genetic/genomic diversity, and is often associated with an increase in phenotypic diversity. Gene (or genome) duplication provides the evolutionary potential for such increase of genomic diversity by neo/subfunctionalisation of one of the gene paralogs, and is often proposed to be related to evolutionary radiations (Ohno 1970; Francino 2005).
References
| Convergent origin and accelerated evolution of vesicle-associated RhoGAP proteins in two unrelated parasitoid wasps | Dominique Colinet, Fanny Cavigliasso, Matthieu Leobold, Appoline Pichon, Serge Urbach, Dominique Cazes, Marine Poullet, Maya Belghazi, Anne-Nathalie Volkoff, Jean-Michel Drezen, Jean-Luc Gatti, and Marylène Poirié | <p>Animal venoms and other protein-based secretions that perform a variety of functions, from predation to defense, are highly complex cocktails of bioactive compounds. Gene duplication, accompanied by modification of the expression and/or functio... | | Evolutionary genomics | Ignacio Bravo | 2023-06-12 11:08:31 | ||
06 Feb 2024

The need of decoding life for taking care of biodiversity and the sustainable use of nature in the Anthropocene - a Faroese perspectiveWhy sequence everything? A raison d’être for the Genome Atlas of Faroese EcologyRecommended by Stephen Richards based on reviews by Tereza Manousaki and 1 anonymous reviewer
When discussing the Earth BioGenome Project with scientists and potential funding agencies, one common question is: why sequence everything? Whether sequencing a subset would be more optimal is not an unreasonable question given what we know about the mathematics of importance and Pareto’s 80:20 principle, that 80% of the benefits can come from 20% of the effort. However, one must remember that this principle is an observation made in hindsight and selecting the most effective 20% of experiments is difficult. As an example, few saw great applied value in comparative genomic analysis of the archaea Haloferax mediterranei, but this enabled the discovery of CRISPR/Cas9 technology (1). When discussing whether or not to sequence all life on our planet, smaller countries such as the Faroe Islands are seldom mentioned.
1 Mojica, F. J., Díez-Villaseñor, C. S., García-Martínez, J. & Soria, E. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J Mol Evol 60, 174-182 (2005). 2 Mikalsen, S-O., Hjøllum, J. í., Salter, I., Djurhuus, A. & Kongsstovu, S. í. The need of decoding life for taking care of biodiversity and the sustainable use of nature in the Anthropocene – a Faroese perspective. EcoEvoRxiv (2024), ver. 3 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.32942/X21S4C | The need of decoding life for taking care of biodiversity and the sustainable use of nature in the Anthropocene - a Faroese perspective | Svein-Ole Mikalsen, Jari í Hjøllum, Ian Salter, Anni Djurhuus, Sunnvør í Kongsstovu | <p>Biodiversity is under pressure, mainly due to human activities and climate change. At the international policy level, it is now recognised that genetic diversity is an important part of biodiversity. The availability of high-quality reference g... | | ERGA, ERGA Pilot, Population genomics, Vertebrates | Stephen Richards | 2023-07-31 16:59:33 | ||
24 Jan 2024

High quality genome assembly of the brown hare (Lepus europaeus) with chromosome-level scaffoldingA high quality reference genome of the brown hareRecommended by Ed Hollox based on reviews by Merce Montoliu-Nerin and 1 anonymous reviewer
The brown hare, or European hare, Lupus europaeus, is a widespread mammal whose natural range spans western Eurasia. At the northern limit of its range, it hybridises with the mountain hare (L. timidis), and humans have introduced it into other continents. It represents a particularly interesting mammal to study for its population genetics, extensive hybridisation zones, and as an invasive species. This study (Michell et al. 2024) has generated a high-quality assembly of a genome from a brown hare from Finland using long PacBio HiFi sequencing reads and Hi-C scaffolding. The contig N50 of this new genome is 43 Mb, and completeness, assessed using BUSCO, is 96.1%. The assembly comprises 23 autosomes, and an X chromosome and Y chromosome, with many chromosomes including telomeric repeats, indicating the high level of completeness of this assembly. While the genome of the mountain hare has previously been assembled, its assembly was based on a short-read shotgun assembly, with the rabbit as a reference genome. The new high-quality brown hare genome assembly allows a direct comparison with the rabbit genome assembly. For example, the assembly addresses the karyotype difference between the hare (n=24) and the rabbit (n=22). Chromosomes 12 and 17 of the hare are equivalent to chromosome 1 of the rabbit, and chromosomes 13 and 16 of the hare are equivalent to chromosome 2 of the rabbit. The new assembly also provides a hare Y-chromosome, as the previous mountain hare genome was from a female. This new genome assembly provides an important foundation for population genetics and evolutionary studies of lagomorphs. References Michell, C., Collins, J., Laine, P. K., Fekete, Z., Tapanainen, R., Wood, J. M. D., Goffart, S., Pohjoismäki, J. L. O. (2024). High quality genome assembly of the brown hare (Lepus europaeus) with chromosome-level scaffolding. bioRxiv, ver. 3 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2023.08.29.555262 | High quality genome assembly of the brown hare (*Lepus europaeus*) with chromosome-level scaffolding | Craig Michell, Joanna Collins, Pia K. Laine, Zsofia Fekete, Riikka Tapanainen, Jonathan M. D. Wood, Steffi Goffart, Jaakko L. O. Pohjoismaki | <p style="text-align: justify;">We present here a high-quality genome assembly of the brown hare (Lepus europaeus Pallas), based on a fibroblast cell line of a male specimen from Liperi, Eastern Finland. This brown hare genome represents the first... | | ERGA Pilot, Vertebrates | Ed Hollox | 2023-10-16 20:46:39 | ||
15 Jan 2024
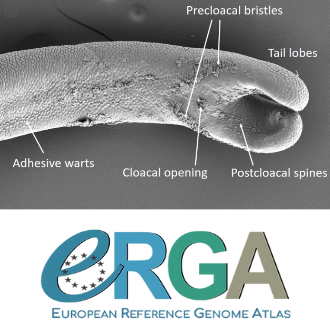
The genome sequence of the Montseny horsehair worm, Gordionus montsenyensis sp. nov., a key resource to investigate Ecdysozoa evolutionEmbarking on a novel journey in Metazoa evolution through the pioneering sequencing of a key underrepresented lineageRecommended by Juan C. Opazo based on reviews by Gonzalo Riadi and 2 anonymous reviewers
Whole genome sequences are revolutionizing our understanding across various biological fields. They not only shed light on the evolution of genetic material but also uncover the genetic basis of phenotypic diversity. The sequencing of underrepresented lineages, such as the one presented in this study, is of critical importance. It is crucial in filling significant gaps in our understanding of Metazoa evolution. Despite the wealth of genome sequences in public databases, it is crucial to acknowledge that some lineages across the Tree of Life are underrepresented or absent. This research represents a significant step towards addressing this imbalance, contributing to the collective knowledge of the global scientific community. In this genome note, as part of the European Reference Genome Atlas pilot effort to generate reference genomes for European biodiversity (Mc Cartney et al. 2023), Klara Eleftheriadi and colleagues (Eleftheriadi et al. 2023) make a significant effort to add a genome sequence of an unrepresented group in the animal Tree of Life. More specifically, they present a taxonomic description and chromosome-level genome assembly of a newly described species of horsehair worm (Gordionus montsenyensis). Their sequence methodology gave rise to an assembly of 396 scaffolds totaling 288 Mb, with an N50 value of 64.4 Mb, where 97% of this assembly is grouped into five pseudochromosomes. The nuclear genome annotation predicted 10,320 protein-coding genes, and they also assembled the circular mitochondrial genome into a 15-kilobase sequence. The selection of a species representing the phylum Nematomorpha, a group of parasitic organisms belonging to the Ecdysozoa lineage, is good, since today, there is only one publicly available genome for this animal phylum (Cunha et al. 2023). Interestingly, this article shows, among other things, that the species analyzed has lost ∼30% of the universal Metazoan genes. Efforts, like the one performed by Eleftheriadi and colleagues, are necessary to gain more insights, for example, on the evolution of this massive gene lost in this group of animals.
Cunha, T. J., de Medeiros, B. A. S, Lord, A., Sørensen, M. V., and Giribet, G. (2023). Rampant Loss of Universal Metazoan Genes Revealed by a Chromosome-Level Genome Assembly of the Parasitic Nematomorpha. Current Biology, 33 (16): 3514–21.e4. https://doi.org/10.1016/j.cub.2023.07.003 Eleftheriadi, K., Guiglielmoni, N., Salces-Ortiz, J., Vargas-Chavez, C., Martínez-Redondo, G. I., Gut, M., Flot, J.-F., Schmidt-Rhaesa, A., and Fernández, R. (2023). The Genome Sequence of the Montseny Horsehair worm, Gordionus montsenyensis sp. Nov., a Key Resource to Investigate Ecdysozoa Evolution. bioRxiv, ver. 3 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2023.06.26.546503 Mc Cartney, A. M., Formenti, G., Mouton, A., De Panis, D., Marins, L. S., Leitão, H. G., Diedericks, G., et al. (2023). The European Reference Genome Atlas: Piloting a Decentralised Approach to Equitable Biodiversity Genomics. bioRxiv. https://doi.org/10.1101/2023.09.25.559365 | The genome sequence of the Montseny horsehair worm, *Gordionus montsenyensis* sp. nov., a key resource to investigate Ecdysozoa evolution | Eleftheriadi Klara, Guiglielmoni Nadège, Salces-Ortiz Judit, Vargas-Chávez Carlos, Martínez-Redondo Gemma I, Gut Marta, Flot Jean François, Schmidt-Rhaesa Andreas, Fernández Rosa | <p>Nematomorpha, also known as Gordiacea or Gordian worms, are a phylum of parasitic organisms that belong to the Ecdysozoa, a clade of invertebrate animals characterized by molting. They are one of the less scientifically studied animal phyla, an... | | ERGA Pilot | Juan C. Opazo | 2023-06-29 10:31:36 | ||
22 Nov 2023
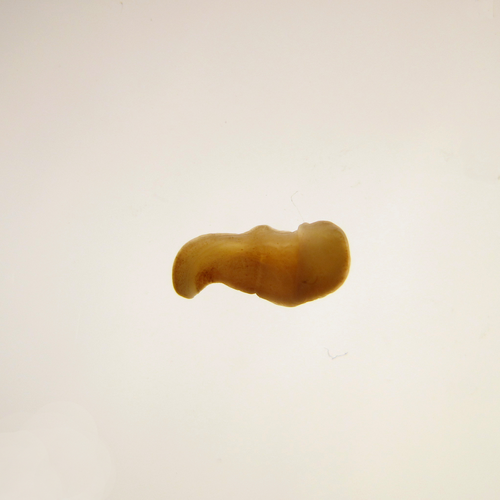
The slow evolving genome of the xenacoelomorph worm Xenoturbella bockiGenomic idiosyncrasies of Xenoturbella bocki: morphologically simple yet genetically complexRecommended by Rosa Fernandez based on reviews by Christopher Laumer and 1 anonymous reviewer
Xenoturbella is a genus of morphologically simple bilaterians inhabiting benthic environments. Until very recently, only one species was known from the genus, Xenoturbella bocki Westblad 1949 [1]. Less than a decade ago, five more species were discovered (X. churro, X. monstrosa, X. profunda, X. hollandorum [2] and X. japonica [3]). These enigmatic animals lack an anus, a coelom, reproductive organs, nephrocytes and a centralized nervous system [1]. The systematic classification of the genus has substantially changed in the last decades, with first being considered as its own phylum (Xenoturbellida) and then being clustered together with acoels and nemertodermatids into the phylum Xenacoelomorpha [4,5]. The phylogenetic position of the xenacoelomorphs has been recalcitrant to resolution, with its position ranging from being the sister group to Nephrozoa (ie, protostomes and deuterostomes [6]) to the sister group to Ambulacraria (ie, Hemichordata and Echinodermata) in a clade called Xenambulacraria [4]. Recent studies based on expanded datasets and more refined analyses support either topology [7,8]. Either way, it is clear that additional studies on Xenoturbella could provide important insights into the origins of bilaterian traits such as the anus, the nephrons and the evolution of a centralized nervous system.
In any case, we are approaching a qualitative jump in how we understand phylogenomics thanks to efforts derived from the availability of chromosome-level genome assemblies for a growing number of species. Exciting times are ahead for us, evolutionary biologists, to explore what high-quality genomes - in combination with multiomics datasets - will reveal about animal evolution. I am personally really looking forward to it. References 1. Westblad E. (1949). Xenoturbella bocki n.g., n.sp., a peculiar, primitive Turbellarian type. Arkiv för Zoologi 1, 3-29 (1949). 2. Rouse, G. W., Wilson, N. G., Carvajal, J. I. & Vrijenhoek, R. C. New deep-sea species of Xenoturbella and the position of Xenacoelomorpha. Nature 530, 94–97 (2016). https://doi.org/10.1038/nature16545 3. Nakano, H. et al. Correction to: A new species of Xenoturbella from the western Pacific Ocean and the evolution of Xenoturbella. BMC Evol. Biol. 18, 1–2 (2018). https://doi.org/10.1186/s12862-018-1190-5 4. Philippe, H. et al. Acoelomorph flatworms are deuterostomes related to Xenoturbella. Nature 470, 255–258 (2011). https://doi.org/10.1038/nature09676 5. Hejnol, A. et al. Assessing the root of bilaterian animals with scalable phylogenomic methods. Proc. Biol. Sci. 276, 4261–4270 (2009). https://doi.org/10.1098/rspb.2009.0896 6. Cannon, J. T. et al. Xenacoelomorpha is the sister group to Nephrozoa. Nature 530, 89–93 (2016). https://doi.org/10.1038/nature16520 7. Laumer, C. E. et al. Revisiting metazoan phylogeny with genomic sampling of all phyla. Proc. Biol. Sci. 286, 20190831 (2019). https://doi.org/10.1098/rspb.2019.0831 8. Philippe, H. et al. Mitigating anticipated effects of systematic errors supports sister-group relationship between Xenacoelomorpha and Ambulacraria. Curr. Biol. 29, 1818–1826.e6 (2019). https://doi.org/10.1016/j.cub.2019.04.009 9. Schiffer, P. H., Natsidis, P., Leite D. J., Robertson, H., Lapraz, F., Marlétaz, F., Fromm, B., Baudry, L., Simpson, F., Høye, E., Zakrzewski, A-C., Kapli, P., Hoff, K. J., Mueller, S., Marbouty, M., Marlow, H., Copley, R. R., Koszul, R., Sarkies, P. & Telford, M .J. The slow evolving genome of the xenacoelomorph worm Xenoturbella bocki. bioRxiv (2023), ver. 4 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2022.06.24.497508 10. Suga, H. et al. The Capsaspora genome reveals a complex unicellular prehistory of animals. Nat. Commun. 4, 2325 (2013). https://doi.org/10.1038/ncomms3325 11. Fernández, R. & Gabaldón, T. Gene gain and loss across the metazoan tree of life. Nat Ecol Evol 4, 524–533 (2020). https://doi.org/10.1038/s41559-019-1069-x | The slow evolving genome of the xenacoelomorph worm *Xenoturbella bocki* | Philipp H. Schiffer, Paschalis Natsidis, Daniel J. Leite, Helen Robertson, François Lapraz, Ferdinand Marlétaz, Bastian Fromm, Liam Baudry, Fraser Simpson, Eirik Høye, Anne-C. Zakrzewski, Paschalia Kapli, Katharina J. Hoff, Steven Mueller, Martial... | <p style="text-align: justify;">The evolutionary origins of Bilateria remain enigmatic. One of the more enduring proposals highlights similarities between a cnidarian-like planula larva and simple acoel-like flatworms. This idea is based in part o... | | Evolutionary genomics | Rosa Fernandez | 2022-11-01 12:31:53 | ||
20 Nov 2023
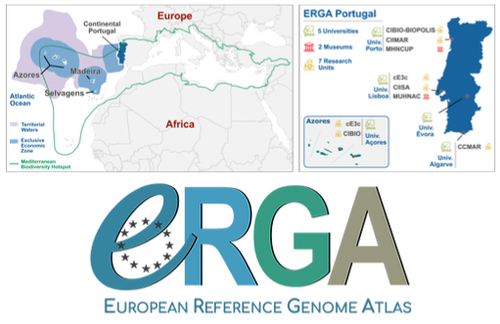
Building a Portuguese Coalition for Biodiversity GenomicsThe Portuguese genomics community teams up with iconic species to understand the destruction of biodiversityRecommended by Fernando Racimo based on reviews by Svein-Ole Mikalsen and 1 anonymous reviewerThis manuscript describes the ongoing work and plans of Biogenome Portugal: a new network of researchers in the Portuguese biodiversity genomics community. The aims of this network are to jointly train scientists in ecology and evolution, generate new knowledge and understanding of Portuguese biodiversity, and better engage with the public and with international researchers, so as to advance conservation efforts in the region. In collaboration across disciplines and institutions, they are also contributing to the European Reference Genome Atlas (ERGA): a massive scientific effort, seeking to eventually produce reference-quality genomes for all species in the European continent (Mc Cartney et al. 2023). The manuscript centers around six iconic and/or severely threatened species, whose range extends across parts of what is today considered Portuguese territory. Via the Portugal chapter of ERGA (ERGA-Portugal), the researchers will generate high-quality genome sequences from these species. The species are the Iberian hare, the Azores laurel, the Black wheatear, the Portuguese crowberry, the Cave ground beetle and the Iberian minnowcarp. In ignorance of human-made political borders, some of these species also occupy large parts of the rest of the Iberian peninsula, highlighting the importance of transnational collaboration in biodiversity efforts. The researchers extracted samples from members of each of these species, and are building reference genome sequences from them. In some cases, these sequences will also be co-analyzed with additional population genomic data from the same species or genetic data from cohabiting species. The researchers aim to answer a variety of ecological and evolutionary questions using this information, including how genetic diversity is being affected by the destruction of their habitat, and how they are being forced to adapt as a consequence of the climate emergency. The authors did a very good job in providing a justification for the choice of pilot species, a thorough methodological overview of current work, and well thought-out plans for future analyses once the genome sequences are available for study. The authors also describe plans for networking and training activities to foster a well-connected Portuguese biodiversity genomics community. Applying a genomic analysis lens is important for understanding the ever faster process of devastation of our natural world. Governments and corporations around the globe are destroying nature at ever larger scales (Diaz et al. 2019). They are also destabilizing the climatic conditions on which life has existed for thousands of years (Trisos et al. 2020). Thus, genetic diversity is decreasing faster than ever in human history, even when it comes to non-threatened species (Exposito-Alonso et al. 2022), and these decreases are disrupting ecological processes worldwide (Richardson et al. 2023). This, in turn, is threatening the conditions on which the stability of our societies rest (Gardner and Bullock 2021). The efforts of Biogenome Portal and ERGA-Portugal will go a long way in helping us understand in greater detail how this process is unfolding in Portuguese territories.
References Díaz, Sandra, et al. "Pervasive human-driven decline of life on Earth points to the need for transformative change." Science 366.6471 (2019): eaax3100. https://doi.org/10.1126/science.aax3100 Exposito-Alonso, Moises, et al. "Genetic diversity loss in the Anthropocene." Science 377.6613 (2022): 1431-1435. https://doi.org/10.1126/science.abn5642 Gardner, Charlie J., and James M. Bullock. "In the climate emergency, conservation must become survival ecology." Frontiers in Conservation Science 2 (2021): 659912. https://doi.org/10.3389/fcosc.2021.659912 Mc Cartney, Ann M., et al. "The European Reference Genome Atlas: piloting a decentralised approach to equitable biodiversity genomics." bioRxiv (2023): 2023-09, ver. 2 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.32942/X20W3Q Richardson, Katherine, et al. "Earth beyond six of nine planetary boundaries." Science Advances 9.37 (2023): eadh2458. https://doi.org/10.1126/sciadv.adh2458 Trisos, Christopher H., Cory Merow, and Alex L. Pigot. "The projected timing of abrupt ecological disruption from climate change." Nature 580.7804 (2020): 496-501. https://doi.org/10.1038/s41586-020-2189-9 | Building a Portuguese Coalition for Biodiversity Genomics | João Pedro Marques, Paulo Célio Alves, Isabel R. Amorim, Ricardo J. Lopes, Mónica Moura, Gene Meyers, Manuela Sim-Sim, Carla Sousa-Santos, Maria Judite Alves, Paulo AV Borges, Thomas Brown, Miguel Carneiro, Carlos Carrapato, Luís Ceríaco, Claudio ... | <p style="text-align: justify;">The diverse physiography of the Portuguese land and marine territory, spanning from continental Europe to the Atlantic archipelagos, has made it an important repository of biodiversity throughout the Pleistocene gla... | | ERGA, ERGA Pilot | Fernando Racimo | 2023-07-14 11:24:22 | ||
14 Sep 2023
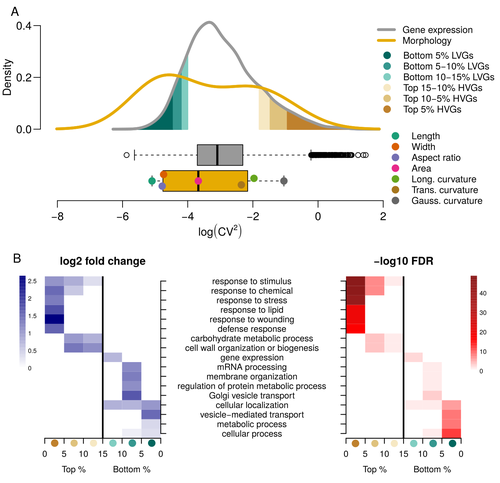
Expression of cell-wall related genes is highly variable and correlates with sepal morphologyThe same but different: How small scale hidden variations can have large effectsRecommended by Francois Sabot based on reviews by Sandra Corjito and 1 anonymous reviewer
For ages, we considered only single genes, or just a few, in order to understand the relationship between phenotype and genotype in response to environmental challenges. Recently, the use of meaningful groups of genes, e.g. gene regulatory networks, or modules of co-expression, allowed scientists to have a larger view of gene regulation. However, all these findings were based on contrasted genotypes, e.g. between wild-types and mutants, as the implicit assumption often made is that there is little transcriptomic variability within the same genotype context. Hartasànchez and collaborators (2023) decided to challenge both views: they used a single genotype instead of two, the famous A. thaliana Col0, and numerous plants, and considered whole gene networks related to sepal morphology and its variations. They used a clever approach, combining high-level phenotyping and gene expression to better understand phenomena and regulations underlying sepal morphologies. Using multiple controls, they showed that basic variations in the expression of genes related to the cell wall regulation, as well as the ones involved in chloroplast metabolism, influenced the global transcriptomic pattern observed in sepal while being in near-identical genetic background and controlling for all other experimental conditions. The paper of Hartasànchez et al. is thus a tremendous call for humility in biology, as we saw in their work that we just understand the gross machinery. However, the Devil is in the details: understanding those very small variations that may have a large influence on phenotypes, and thus on local adaptation to environmental challenges, is of great importance in these times of climatic changes. References Hartasánchez DA, Kiss A, Battu V, Soraru C, Delgado-Vaquera A, Massinon F, Brasó-Vives M, Mollier C, Martin-Magniette M-L, Boudaoud A, Monéger F. 2023. Expression of cell-wall related genes is highly variable and correlates with sepal morphology. bioRxiv, ver. 4, peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2022.04.26.489498 | Expression of cell-wall related genes is highly variable and correlates with sepal morphology | Diego A. Hartasánchez, Annamaria Kiss, Virginie Battu, Charline Soraru, Abigail Delgado-Vaquera, Florian Massinon, Marina Brasó-Vives, Corentin Mollier, Marie-Laure Martin-Magniette, Arezki Boudaoud, Françoise Monéger | <p style="text-align: justify;">Control of organ morphology is a fundamental feature of living organisms. There is, however, observable variation in organ size and shape within a given genotype. Taking the sepal of Arabidopsis as a model, we inves... | | Bioinformatics, Epigenomics, Plants | Francois Sabot | 2023-03-14 19:10:15 |





