
RAMOS-ONSINS Sebastian Ernesto
- Plant and Animal Genomics, Center for Research in Agricultural Genomics, Cerdanyola del Valles, Spain
- Bioinformatics, Evolutionary genomics, Plants, Population genomics
- recommender
Recommendations: 3
Reviews: 0
Recommendations: 3
Estimating allele frequencies, ancestry proportions and genotype likelihoods in the presence of mapping bias
A novel genotype likelihood-based method to reduce mapping bias in low-coverage and ancient DNA studies
Recommended by Sebastian Ernesto Ramos-Onsins based on reviews by Maxime Lefebvre, Michael Westbury and Adrien OlivaThe study of genomic variability within and between populations, as well as among species, relies on comparative analyses of homologous positions—sites that share a common evolutionary origin. Homology is inferred through sequence similarity (Reeck et al. 1987). However, the ability to detect homologous regions can be compromised when sequence mismatches accumulate due to mutations, especially when analyzing short DNA fragments, as in short-read sequencing (Li et al. 2008). In the genomic era, accurately mapping homologous DNA fragments to a reference genome is essential for obtaining precise estimates of genetic variability and evolutionary inferences (e.g., Li et al. 2008; Ellegren 2014). However, short-read, high-throughput sequencing often introduces mapping bias, disproportionately favoring the reference allele. This bias distorts allele frequency estimates, ancestry proportions, and genotype likelihoods, impacting downstream analyses (e.g., Günther & Nettelblad 2019; Martiniano et al. 2020).
Mapping bias is particularly problematic in ancient DNA studies, where post-mortem damage exacerbates sequencing errors. DNA fragmentation limits read length, while deamination, causing G to A and C to U transitions, increases mismatches and further complicates homology identification (Dabney & Pääbo 2013). These degradation processes contribute to the misidentification of true variants, confounding evolutionary inferences. Various strategies have been developed to mitigate mapping bias, including the commonly used approach, called pseudo-haploid data, that randomly picks a single read at each analyzed position for each individual, thereby retaining a single allele at each polymorphic site (Günther & Nettelblad 2019; Barlow et al. 2020).
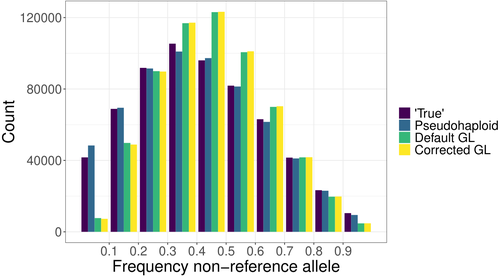
Günther et al. (2025) introduce a novel method to correct mapping bias using a genotype likelihood-based approach, incorporating a mapping bias ratio to adjust for reference allele overrepresentation. The method specifically targets known single nucleotide polymorphisms (SNPs) because in population genomic analysis of ancient DNA data, low coverage and post-mortem damage often hinder the ability to identify novel SNPs in most individuals. The analysis focuses on DNA fragmentation, assuming that deamination effects are minimal when considering ascertained SNPs. The proposed method was compared against existing approaches, including pseudo-haploid data and standard genotype likelihood-based probabilistic methods. The evaluation was performed using both empirical and simulated data. For empirical data, low-coverage sequencing data from the 1000 Genomes Project (Finnish in Finland, Japanese in Tokyo, Yoruba in Ibadan, Nigeria populations) was analyzed, while for simulated data, ancient DNA-like datasets were generated using ms-prime (Kelleher et al. 2016), modeling different sequencing depths, divergence times, and reference genome choices. The study assesses the impact of mapping bias on the ratio of reference versus non-reference allele mapping, the accuracy of SNP allele frequency estimates relative to true frequencies, the deviation and variance between estimated and true allele frequencies, population differentiation and the estimation of admixture proportions using supervised and unsupervised methods, considering both genotype likelihoods and genotype calls.
Günther et al. (2025) bring to light that all methods analyzed exhibit minor but systematic reference allele bias. The new corrected genotype likelihood method outperforms the standard genotype likelihood approach in correlating with true allele frequencies, although the pseudo-haploid method still provides the most accurate estimates. Mapping bias also affects ancestry estimation, leading to admixture proportion errors of up to 4%, though this effect is smaller than the 10% discrepancy observed across different inference methods.
The work performed by Günther et al. (2025) provides a rigorous and innovative evaluation of mapping bias in the context of ascertained SNPs, introducing a probabilistic approach that improves bias correction. Unlike non-probabilistic methods such as pseudo-haploid data, the genotype likelihood framework leverages all sequencing reads for each analyzed SNP, and can incorporate additional bias corrections, enhancing its applicability across different sequencing conditions. While probabilistic approaches offer clear advantages in bias correction, they can be less intuitive to interpret compared to traditional genotype calling methods. This study highlights that mapping bias is pervasive across all methods, influencing evolutionary inferences such as selection signals and population differentiation. Although the improvements in allele frequency recovery may seem modest, the genome-wide impact of mapping bias is significant, especially in ancient DNA studies, making bias correction essential for robust evolutionary analyses.
References
Barlow A, Hartmann S, Gonzalez J, Hofreiter M, Paijmans JLA. (2020) Consensify: A method for generating pseudohaploid genome sequences from palaeogenomic datasets with reduced error rates. Genes;11(1):50. https://doi.org/10.3390/genes11010050
Dabney J, Meyer M, Pääbo S. (2013) Ancient DNA damage. Cold Spring Harb Perspect Biol. 5(7):a012567. https://doi.org/10.1101/cshperspect.a012567
Ellegren H. (2014) Genome sequencing and population genomics in non-model organisms. Trends Ecol Evol. 29(1):51-63. https://doi.org/10.1016/j.tree.2013.09.008
Günther T, Nettelblad C. (2019) The presence and impact of reference bias on population genomic studies of prehistoric human populations. PLoS Genet.15(7):e1008302. https://doi.org/10.1371/journal.pgen.1008302
Günther T., Goldberg A., Schraiber J. G. (2025) Estimating allele frequencies, ancestry proportions and genotype likelihoods in the presence of mapping bias. bioRxiv, ver. 5 peer-reviewed and recommended by PCI Genomics https://doi.org/10.1101/2024.07.01.601500
Kelleher J., Etheridge A. M., McVean G. (2016) Efficient coalescent simulation and genealogical analysis for large sample sizes. PLoS computational biology, 12(5):e1004842. https://doi.org/10.1371/journal.pcbi.1004842
Li H, Ruan J, Durbin R. (2008) Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 18(11):1851-8. https://doi.org/10.1101/gr.078212.108
Reeck GR, de Haën C, Teller DC, Doolittle RF, Fitch WM, Dickerson RE, et al. (1987) "Homology" in proteins and nucleic acids: a terminology muddle and a way out of it. Cell. 50 (5): 667. https://doi.org/10.1016/0092-8674(87)90322-9
Performance and limitations of linkage-disequilibrium-based methods for inferring the genomic landscape of recombination and detecting hotspots: a simulation study
How to interpret the inference of recombination landscapes on methods based on linkage disequilibrium?
Recommended by Sebastian Ernesto Ramos-Onsins based on reviews by 2 anonymous reviewersData interpretation depends on previously established and validated tools, designed for a specific type of data. These methods, however, are usually based on simple models with validity subject to a set of theoretical parameterized conditions and data types. Accordingly, the tool developers provide the potential users with guidelines for data interpretations within the tools’ limitation. Nevertheless, once the methodology is accepted by the community, it is employed in a large variety of empirical studies outside of the method’s original scope or that typically depart from the standard models used for its design, thus potentially leading to the wrong interpretation of the results.
Numerous empirical studies inferred recombination rates across genomes, detecting hotspots of recombination and comparing related species (e.g., Shanfelter et al. 2019, Spence and Song 2019). These studies used indirect methodologies based on the signals that recombination left in the genome, such as linkage disequilibrium and the patterns of haplotype segregation (e.g.,Chan et al. 2012). The conclusions from these analyses have been used, for example, to interpret the evolution of the chromosomal structure or the evolution of recombination among closely related species.
Indirect methods have the advantage of collecting a large quantity of recombination events, and thus have a better resolution than direct methods (which only detect the few recombination events occurring at that time). On the other hand, indirect methods are affected by many different evolutionary events, such as demographic changes and selection. Indeed, the inference of recombination levels across the genome has not been studied accurately in non-standard conditions. Linkage disequilibrium is affected by several factors that can modify the recombination inference, such as demographic history, events of selection, population size, and mutation rate, but is also related to the size of the studied sample, and other technical parameters defined for each specific methodology.
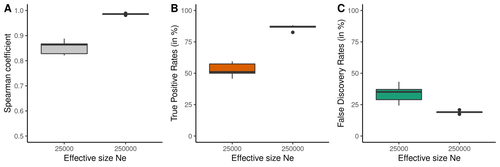
Raynaud et al (2023) analyzed the reliability of the recombination rate inference when considering the violation of several standard assumptions (evolutionary and methodological) in one of the most popular families of methods based on LDhat (McVean et al. 2004), specifically its improved version, LDhelmet (Chan et al. 2012). These methods cover around 70 % of the studies that infer recombination rates. The authors used recombination maps, obtained from empirical studies on humans, and included hotspots, to perform a detailed simulation study of the capacity of this methodology to correctly infer the pattern of recombination and the location of these hotspots. Correlations between the real, and inferred values from simulations were obtained, as well as several rates, such as the true positive and false discovery rate to detect hotspots.
The authors of this work send a message of caution to researchers that are applying this methodology to interpret data from the inference of recombination landscapes and the location of hotspots. The inference of recombination landscapes and hotspots can differ considerably even in standard model conditions. In addition, demographic processes, like bottleneck or admixture, but also the level of population size and mutation rates, can substantially affect the estimation accuracy of the level of recombination and the location of hotspots. Indeed, the inference of the location of hotspots in simulated data with the same landscape, can be very imprecise when standard assumptions are violated or not considered. These effects may lead to incorrect interpretations, for example about the conservation of recombination maps between closely related species. Finally, Raynaud et al (2023) included a useful guide with advice on how to obtain accurate recombination estimations with methods based on linkage disequilibrium, also emphasizing the limitations of such approaches.
REFERENCES
Chan AH, Jenkins PA, Song YS (2012) Genome-Wide Fine-Scale Recombination Rate Variation in Drosophila melanogaster. PLOS Genetics, 8, e1003090. https://doi.org/10.1371/journal.pgen.1003090
McVean GAT, Myers SR, Hunt S, Deloukas P, Bentley DR, Donnelly P (2004) The Fine-Scale Structure of Recombination Rate Variation in the Human Genome. Science, 304, 581–584. https://doi.org/10.1126/science.1092500
Raynaud M, Gagnaire P-A, Galtier N (2023) Performance and limitations of linkage-disequilibrium-based methods for inferring the genomic landscape of recombination and detecting hotspots: a simulation study. bioRxiv, 2022.03.30.486352, ver. 2 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2022.03.30.486352
Spence JP, Song YS (2019) Inference and analysis of population-specific fine-scale recombination maps across 26 diverse human populations. Science Advances, 5, eaaw9206. https://doi.org/10.1126/sciadv.aaw9206
An evaluation of pool-sequencing transcriptome-based exon capture for population genomics in non-model species
Assessing a novel sequencing-based approach for population genomics in non-model species
Recommended by Thomas Derrien and Sebastian Ernesto Ramos-Onsins based on reviews by Valentin Wucher and 1 anonymous reviewerDeveloping new sequencing and bioinformatic strategies for non-model species is of great interest in many applications, such as phylogenetic studies of diverse related species, but also for studies in population genomics, where a relatively large number of individuals is necessary. Different approaches have been developed and used in these last two decades, such as RAD-Seq (e.g., Miller et al. 2007), exome sequencing (e.g., Teer and Mullikin 2010) and other genome reduced representation methods that avoid the use of a good reference and well annotated genome (reviewed at Davey et al. 2011). However, population genomics studies require the analysis of numerous individuals, which makes the studies still expensive. Pooling samples was thought as an inexpensive strategy to obtain estimates of variability and other related to the frequency spectrum, thus allowing the study of variability at population level (e.g., Van Tassell et al. 2008), although the major drawback was the loss of information related to the linkage of the variants. In addition, population analysis using all these sequencing strategies require statistical and empirical validations that are not always fully performed. A number of studies aiming to obtain unbiased estimates of variability using reduced representation libraries and/or with pooled data have been performed (e.g., Futschik and Schlötterer 2010, Gautier et al. 2013, Ferretti et al. 2013, Lynch et al. 2014), as well as validation of new sequencing methods for population genetic analyses (e.g., Gautier et al. 2013, Nevado et al. 2014). Nevertheless, empirical validation using both pooled and individual experimental approaches combined with different bioinformatic methods has not been always performed.
Here, Deleury et al. (2020) proposed an efficient and elegant way of quantifying the single-nucleotide polymorphisms (SNPs) of exon-derived sequences in a non-model species (i.e. for which no reference genome sequence is available) at the population level scale. They also designed a new procedure to capture exon-derived sequences based on a reference transcriptome. In addition, they were able to make predictions of intron-exon boundaries for de novo transcripts based on the decay of read depth at the ends of the coding regions.
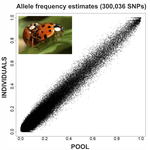
Based on theoretical predictions (Gautier et al. 2013), Deleury et al. (2020) designed a procedure to test the accuracy of variant allele frequencies (AFs) with pooled samples, in a reduced genome-sequence library made with transcriptome regions, and additionally testing the effects of new bioinformatic methods in contrast to standardized methods. They applied their strategy on the non-model species Asian ladybird (Harmonia axyridis), for which a draft genome is available, thereby allowing them to benchmark their method with regard to a traditional mapping-based approach. Based on species-specific de novo transcriptomes, they designed capture probes which are then used to call SNPx and then compared the resulting SNP AFs at the individual (multiplexed) versus population (pooled) levels. Interestingly, they showed that SNP AFs in the pool sequencing strategy nicely correlate with the individual ones but obviously in a cost-effective way. Studies of population genomics for non-model species have usually limited budgets. The number of individuals required for population genomics analysis multiply the costs of the project, making pooling samples an interesting option. Furthermore, the use of pool sequencing is not always a choice, as many organisms are too small and/or individuals are too sticked each other to be individually sequenced (e.g., Choquet et al. 2019, Kurland et al. 2019). In addition, the study of a reduced section of the genome is cheaper and often sufficient for a number of population genetic questions, such as the understanding of general demographic events, or the estimation of the effects of positive and/or negative selection at functional coding regions. Studies on population genomics of non-model species have many applications in related fields, such as conservation genetics, control of invasive species, etc. The work of Deleury et al. (2020) is an elegant contribution to the assessment and validation of new methodologies used for the analysis of genome variations at the intra-population variability level, highlighting straight bioinformatic and reliable sequencing methods for population genomics studies.
References
[1] Choquet et al. (2019). Towards population genomics in non-model species with large genomes: a case study of the marine zooplankton Calanus finmarchicus. Royal Society open science, 6(2), 180608. doi: https://doi.org/10.1098/rsos.180608
[2] Davey, J. W., Hohenlohe, P. A., Etter, P. D., Boone, J. Q., Catchen, J. M. and Blaxter, M. L. (2011). Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nature Reviews Genetics, 12(7), 499-510. doi: https://doi.org/10.1038/nrg3012
[3] Deleury, E., Guillemaud, T., Blin, A. and Lombaert, E. (2020) An evaluation of pool-sequencing transcriptome-based exon capture for population genomics in non-model species. bioRxiv, 10.1101/583534, ver. 7 peer-reviewed and recommended by PCI Genomics. https://doi.org/10.1101/583534
[4] Ferretti, L., Ramos‐Onsins, S. E. and Pérez‐Enciso, M. (2013). Population genomics from pool sequencing. Molecular ecology, 22(22), 5561-5576. doi: https://doi.org/10.1111/mec.12522
[5] Futschik, A. and Schlötterer, C. (2010). Massively parallel sequencing of pooled DNA samples—the next generation of molecular markers. Genetics, 186 (1), 207-218. doi: https://doi.org/10.1534/genetics.110.114397
[6] Gautier et al. (2013). Estimation of population allele frequencies from next‐generation sequencing data: pool‐versus individual‐based genotyping. Molecular Ecology, 22(14), 3766-3779. doi: https://doi.org/10.1111/mec.12360
[7] Kurland et al. (2019). Exploring a Pool‐seq‐only approach for gaining population genomic insights in nonmodel species. Ecology and evolution, 9(19), 11448-11463. doi: https://doi.org/10.1002/ece3.5646
[8] Lynch, M., Bost, D., Wilson, S., Maruki, T. and Harrison, S. (2014). Population-genetic inference from pooled-sequencing data. Genome biology and evolution, 6(5), 1210-1218. doi: https://doi.org/10.1093/gbe/evu085
[9] Miller, M. R., Dunham, J. P., Amores, A., Cresko, W. A. and Johnson, E. A. (2007). Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers. Genome research, 17(2), 240-248. doi: https://doi.org/10.1101%2Fgr.5681207
[10] Nevado, B., Ramos‐Onsins, S. E. and Perez‐Enciso, M. (2014). Resequencing studies of nonmodel organisms using closely related reference genomes: optimal experimental designs and bioinformatics approaches for population genomics. Molecular ecology, 23(7), 1764-1779. doi: https://doi.org/10.1111/mec.12693
[11] Teer, J. K. and Mullikin, J. C. (2010). Exome sequencing: the sweet spot before whole genomes. Human molecular genetics, 19(R2), R145-R151. doi: https://doi.org/10.1093/hmg/ddq333
[12] Van Tassell et al. (2008). SNP discovery and allele frequency estimation by deep sequencing of reduced representation libraries. Nature methods, 5(3), 247-252. doi: https://doi.org/10.1038/nmeth.1185