
SABOT Francois
- DIADE Unit, IRD, Montpellier, France
- Bioinformatics, Evolutionary genomics, Plants, Population genomics, Structural genomics, Viruses and transposable elements
- recommender
Recommendations: 5
Reviews: 0
Recommendations: 5
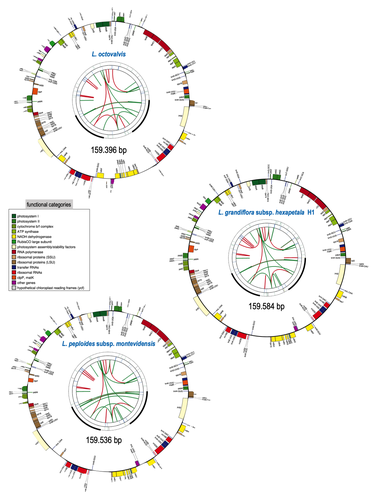
Sequencing, de novo assembly of Ludwigia plastomes, and comparative analysis within the Onagraceae family
Onagre, monster, invasion and genetics
Recommended by Francois Sabot based on reviews by 2 anonymous reviewersThe first time I heard of ”onagres” in French was when I was a teenager, through the books of Pierre Bordage as fantastic monsters, or through historical games as Roman siege weapons (onagers). At this time, I was far from imagining that “onagre” also refers to a very large flowering plant family, as it is the French term for evening primroses.
In this family, the genus Ludwigia comprises species that are invasive (resembling in that way the ancient armies using onagers to invade cities) in aquatic environments, degrading ecosystems already fragilized by human activities. To counteract this phenomenon, it is of high importance to understand their propagation of these species. However, the knowledge about their genetics and diversity is very scarce, and thus tracking their dispersal using genetic information is complicated, and in fact almost impossible.
Barloy-Hubler et al. (2024) proposed in the present manuscript a new set of chloroplastic genomes from two of these species, Ludwigia grandiflora subsp. hexapetala and Ludwigia peploides subsp. montevidensis, and compared them to the published chloroplastic genome of Ludwigia octovalis. They explored the possibility of assembling these genomes relying solely on short reads and showed that long reads were necessary to obtain an almost complete assembly for these plastid genomes. In addition, through this approach, they detected two haplotypes in Ludwigia grandiflora subsp. hexapetala as compared to one in a short-read assembly. This highlights the need for long reads data to assess the structure and diversity of chloroplastic genomes. The authors were also able to clarify the phylogeny of the genus Ludwigia. Finally, they identified multiple potential single nucleotide polymorphisms and simple sequence repeats for future evaluation of diversity and dispersal of those invasive species.
This analysis, while appearing more technical than biological at first glance, is in fact of high importance for the understanding of ecology and preservation of fragile ecosystems, such as the European watersheds. Indeed, new scientific results and insights are generally linked to a reevaluation of previously analyzed data or samples through new technologies, and this paper is a quite clever example of that matter.
References
Barloy-Hubler F, Gac A-LL, Boury C, Guichoux E, Barloy D (2024) Sequencing, de novo assembly of Ludwigia plastomes, and comparative analysis within the Onagraceae family. bioRxiv, ver. 5 peer-reviewed and recommended by PCI Genomics. https://doi.org/10.1101/2023.10.20.563230
Bordage, P (1993) Les Guerriers du Silence, L'Atalante, ISBN 9782905158697
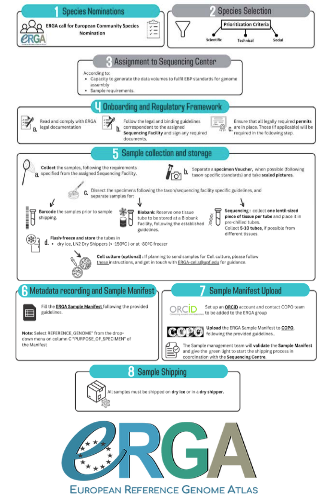
Contextualising samples: Supporting reference genomes of European biodiversity through sample and associated metadata collection
To avoid biases and to be FAIR, we need to CARE and share biodiversity metadata
Recommended by Francois Sabot based on reviews by Julian Osuji and 1 anonymous reviewerBöhne et al. (2024) do not present a classical scientific paper per se but a report on how the European Reference Genome Atlas (ERGA) aims to deal with sampling and sample information, i.e. metadata.
As the goal of ERGA is to provide an almost fully representative set of reference genomes representative of European biodiversity to serve many research areas in biology, they have to be really exhaustive. In this regard, in addition to providing sample metadata recording guidelines, they also discuss the biases existing in sampling and sequencing projects.
The first task for such a project is to be sure that the data they generate will be usable and available in the future (“[in] perpetuity", Böhne et al. 2024). The authors deployed a very efficient pipeline for conserving information on sampling: location, physical information, copies of tissues and of DNA, shipping, legal/ethical aspects regarding the Nagoya Protocol, etc., alongside a best-practice manual. This effort is linked to practical guides for the DNA extraction of specific taxa. More generally, these details enable “Findable, Accessible, Interoperable, and Reusable” (FAIR) principles (Wilkinson et al. 2016) to be followed.
An important aspect of this paper, in addition to practical points, is the reflection upon the different biases inherent to the choice of sequenced samples. Acknowledging their own biases with regards to DNA extraction protocol efficiency, small genome size choice, as well as the availability of material (Nagoya Protocol aspects) and material transfer efficiency, the authors recommend in the future to not survey biodiversity by selecting one’s favorite samples or species, but also considering "orphan" taxa. Some of these "orphan" taxonomic groups belong to non-arthropod invertebrates but internal disparities are also prominent within other taxa. Finally, the implementation of the "Collective benefit, Authority to control, Responsibility, and Ethics" (CARE) principles (Carroll et al. 2021) will allow Indigenous rights to be considered when prioritizing samples, and to enable their "knowledge systems to permeate throughout the process of reference genome production and beyond" (Böhne et al. 2024).
Last, but not least, as ERGA, including its Sampling and Sample Processing committee, is a large collective effort, it is very refreshing to read a paper starting with the acknowledgements and the roles of each member.
References
Böhne A, Fernández R, Leonard JA, McCartney AM, McTaggart S, Melo-Ferreira J, Monteiro R, Oomen RA, Pettersson OV, Struck TH (2024) Contextualising samples: Supporting reference genomes of European biodiversity through sample and associated metadata collection. bioRxiv, ver. 3 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2023.06.28.546652
Carroll SR, Herczog E, Hudson M, Russell K, Stall S (2021) Operationalizing the CARE and FAIR Principles for Indigenous data futures. Scientific Data, 8, 108. https://doi.org/10.1038/s41597-021-00892-0
Wilkinson MD, Dumontier M, Aalbersberg IjJ, Appleton G, Axton M, Baak A, Blomberg N, Boiten J-W, da Silva Santos LB, Bourne PE, Bouwman J, Brookes AJ, Clark T, Crosas M, Dillo I, Dumon O, Edmunds S, Evelo CT, Finkers R, Gonzalez-Beltran A, Gray AJG, Groth P, Goble C, Grethe JS, Heringa J, ’t Hoen PAC, Hooft R, Kuhn T, Kok R, Kok J, Lusher SJ, Martone ME, Mons A, Packer AL, Persson B, Rocca-Serra P, Roos M, van Schaik R, Sansone S-A, Schultes E, Sengstag T, Slater T, Strawn G, Swertz MA, Thompson M, van der Lei J, van Mulligen E, Velterop J, Waagmeester A, Wittenburg P, Wolstencroft K, Zhao J, Mons B (2016) The FAIR Guiding Principles for scientific data management and stewardship. Scientific Data, 3, 160018. https://doi.org/10.1038/sdata.2016.18
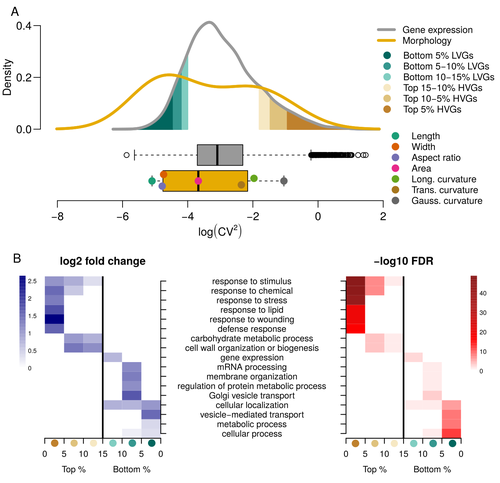
Expression of cell-wall related genes is highly variable and correlates with sepal morphology
The same but different: How small scale hidden variations can have large effects
Recommended by Francois Sabot based on reviews by Sandra Corjito and 1 anonymous reviewerFor ages, we considered only single genes, or just a few, in order to understand the relationship between phenotype and genotype in response to environmental challenges. Recently, the use of meaningful groups of genes, e.g. gene regulatory networks, or modules of co-expression, allowed scientists to have a larger view of gene regulation. However, all these findings were based on contrasted genotypes, e.g. between wild-types and mutants, as the implicit assumption often made is that there is little transcriptomic variability within the same genotype context.
Hartasànchez and collaborators (2023) decided to challenge both views: they used a single genotype instead of two, the famous A. thaliana Col0, and numerous plants, and considered whole gene networks related to sepal morphology and its variations. They used a clever approach, combining high-level phenotyping and gene expression to better understand phenomena and regulations underlying sepal morphologies. Using multiple controls, they showed that basic variations in the expression of genes related to the cell wall regulation, as well as the ones involved in chloroplast metabolism, influenced the global transcriptomic pattern observed in sepal while being in near-identical genetic background and controlling for all other experimental conditions.
The paper of Hartasànchez et al. is thus a tremendous call for humility in biology, as we saw in their work that we just understand the gross machinery. However, the Devil is in the details: understanding those very small variations that may have a large influence on phenotypes, and thus on local adaptation to environmental challenges, is of great importance in these times of climatic changes.
References
Hartasánchez DA, Kiss A, Battu V, Soraru C, Delgado-Vaquera A, Massinon F, Brasó-Vives M, Mollier C, Martin-Magniette M-L, Boudaoud A, Monéger F. 2023. Expression of cell-wall related genes is highly variable and correlates with sepal morphology. bioRxiv, ver. 4, peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2022.04.26.489498

A deep dive into genome assemblies of non-vertebrate animals
Diving, and even digging, into the wild jungle of annotation pathways for non-vertebrate animals
Recommended by Francois Sabot based on reviews by Yann Bourgeois, Cécile Monat, Valentina Peona and Benjamin IstaceIn their paper, Guiglielmoni et al. propose we pick up our snorkels and palms and take "A deep dive into genome assemblies of non-vertebrate animals" (1). Indeed, while numerous assembly-related tools were developed and tested for human genomes (or at least vertebrates such as mice), very few were tested on non-vertebrate animals so far. Moreover, most of the benchmarks are aimed at raw assembly tools, and very few offer a guide from raw reads to an almost finished assembly, including quality control and phasing.
This huge and exhaustive review starts with an overview of the current sequencing technologies, followed by the theory of the different approaches for assembly and their implementation. For each approach, the authors present some of the most representative tools, as well as the limits of the approach.
The authors additionally present all the steps required to obtain an almost complete assembly at a chromosome-scale, with all the different technologies currently available for scaffolding, QC, and phasing, and the way these tools can be applied to non-vertebrates animals. Finally, they propose some useful advice on the choice of the different approaches (but not always tools, see below), and advocate for a robust genome database with all information on the way the assembly was obtained.
This review is a very complete one for now and is a very good starting point for any student or scientist interested to start working on genome assembly, from either model or non-model organisms. However, the authors do not provide a list of tools or a benchmark of them as a recommendation. Why? Because such a proposal may be obsolete in less than a year.... Indeed, with the explosion of the 3rd generation of sequencing technology, assembly tools (from different steps) are constantly evolving, and their relative performance increases on a monthly basis. In addition, some tools are really efficient at the time of a review or of an article, but are not further developed later on, and thus will not evolve with the technology. We have all seen it with wonderful tools such as Chiron (2) or TopHat (3), which were very promising ones, but cannot be developed further due to the stop of the project, the end of the contract of the post-doc in charge of the development, or the decision of the developer to switch to another paradigm. Such advice would, therefore, need to be constantly updated.
Thus, the manuscript from Guiglielmoni et al will be an almost intemporal one (up to the next sequencing revolution at last), and as they advocated for a more informed genome database, I think we should consider a rolling benchmarking system (tools, genome and sequence dataset) allowing to keep the performance of the tools up-to-date, and to propose the best set of assembly tools for a given type of genome.
References
1. Guiglielmoni N, Rivera-Vicéns R, Koszul R, Flot J-F (2022) A Deep Dive into Genome Assemblies of Non-vertebrate Animals. Preprints, 2021110170, ver. 3 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.20944/preprints202111.0170
2. Teng H, Cao MD, Hall MB, Duarte T, Wang S, Coin LJM (2018) Chiron: translating nanopore raw signal directly into nucleotide sequence using deep learning. GigaScience, 7, giy037. https://doi.org/10.1093/gigascience/giy037
3. Trapnell C, Pachter L, Salzberg SL (2009) TopHat: discovering splice junctions with RNA-Seq. Bioinformatics, 25, 1105–1111. https://doi.org/10.1093/bioinformatics/btp120
Traces of transposable element in genome dark matter co-opted by flowering gene regulation networks
Using small fragments to discover old TE remnants: the Duster approach empowers the TE detection
Recommended by Francois Sabot based on reviews by Josep Casacuberta and 1 anonymous reviewerTransposable elements are the raw material of the dark matter of the genome, the foundation of the next generation of genes and regulation networks". This sentence could be the essence of the paper of Baud et al. (2021). Transposable elements (TEs) are endogenous mobile genetic elements found in almost all genomes, which were discovered in 1948 by Barbara McClintock (awarded in 1983 the only unshared Medicine Nobel Prize so far). TEs are present everywhere, from a single isolated copy for some elements to more than millions for others, such as Alu. They are founders of major gene lineages (HET-A, TART and telomerases, RAG1/RAG2 proteins from mammals immune system; Diwash et al, 2017), and even of retroviruses (Xiong & Eickbush, 1988). However, most TEs appear as selfish elements that replicate, land in a new genomic region, then start to decay and finally disappear in the midst of the genome, turning into genomic ‘dark matter’ (Vitte et al, 2007). The mutations (single point, deletion, recombination, and so on) that occur during this slow death erase some of their most notable features and signature sequences, rendering them completely unrecognizable after a few million years. Numerous TE detection tools have tried to optimize their detection (Goerner-Potvin & Bourque, 2018), but further improvement is definitely challenging. This is what Baud et al. (2021) accomplished in their paper. They used a simple, elegant and efficient k-mer based approach to find small signatures that, when accumulated, allow identifying very old TEs. Using this method, called Duster, they improved the amount of annotated TEs in the model plant Arabidopsis thaliana by 20%, pushing the part of this genome occupied by TEs up from 40 to almost 50%. They further observed that these very old Duster-specific TEs (i.e., TEs that are only detected by Duster) are, among other properties, close to genes (much more than recent TEs), not targeted by small RNA pathways, and highly associated with conserved regions across the rosid family. In addition, they are highly associated with flowering or stress response genes, and may be involved through exaptation in the evolution of responses to environmental changes. TEs are not just selfish elements: more and more studies have shown their key role in the evolution of their hosts, and tools such as Duster will help us better understand their impact.
References
Baud, A., Wan, M., Nouaud, D., Francillonne, N., Anxolabéhère, D. and Quesneville, H. (2021). Traces of transposable elements in genome dark matter co-opted by flowering gene regulation networks. bioRxiv, 547877, ver. 5 peer-reviewed and recommended by PCI Genomics.doi: https://doi.org/10.1101/547877
Bourque, G., Burns, K.H., Gehring, M. et al. (2018) Ten things you should know about transposable elements. Genome Biology 19:199. doi: https://doi.org/10.1186/s13059-018-1577-z
Goerner-Potvin, P., Bourque, G. Computational tools to unmask transposable elements. Nature Reviews Genetics 19:688–704 (2018) https://doi.org/10.1038/s41576-018-0050-x
Jangam, D., Feschotte, C. and Betrán, E. (2017) Transposable element domestication as an adaptation to evolutionary conflicts. Trends in Genetics 33:817-831. doi: https://doi.org/10.1016/j.tig.2017.07.011
Vitte, C., Panaud, O. and Quesneville, H. (2007) LTR retrotransposons in rice (Oryza sativa, L.): recent burst amplifications followed by rapid DNA loss. BMC Genomics 8:218. doi: https://doi.org/10.1186/1471-2164-8-218
Xiong, Y. and Eickbush, T. H. (1988) Similarity of reverse transcriptase-like sequences of viruses, transposable elements, and mitochondrial introns. Molecular Biology and Evolution 5: 675–690. doi: https://doi.org/10.1093/oxfordjournals.molbev.a040521