
CASTANERA Raúl
- Structure and evolution of plant genomes, Centre For Research in Agricultural Genomics, Cerdanyola del Vallés, Barcelona, Spain
- Functional genomics, Bioinformatics
- recommender
Recommendation: 1
Reviews: 0
Recommendation: 1
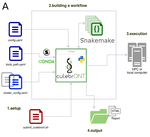
CulebrONT: a streamlined long reads multi-assembler pipeline for prokaryotic and eukaryotic genomes
A flexible and reproducible pipeline for long-read assembly and evaluation
Recommended by Raúl Castanera based on reviews by Benjamin Istace and Valentine MurigneuxThird-generation sequencing has revolutionised de novo genome assembly. Thanks to this technology, genome reference sequences have evolved from fragmented drafts to gapless, telomere-to-telomere genome assemblies. Long reads produced by Oxford Nanopore and PacBio technologies can span structural variants and resolve complex repetitive regions such as centromeres, unlocking previously inaccessible genomic information. Nowadays, many research groups can afford to sequence the genome of their working model using long reads. Nevertheless, genome assembly poses a significant computational challenge. Read length, quality, coverage and genomic features such as repeat content can affect assembly contiguity, accuracy, and completeness in almost unpredictable ways. Consequently, there is no best universal software or protocol for this task. Producing a high-quality assembly requires chaining several tools into pipelines and performing extensive comparisons between the assemblies obtained by different tool combinations to decide which one is the best. This task can be extremely challenging, as the number of tools available rises very rapidly, and thorough benchmarks cannot be updated and published at such a fast pace.
In their paper, Orjuela and collaborators present CulebrONT [1], a universal pipeline that greatly contributes to overcoming these challenges and facilitates long-read genome assembly for all taxonomic groups. CulebrONT incorporates six commonly used assemblers and allows to perform assembly, circularization (if needed), polishing, and evaluation in a simple framework. One important aspect of CulebrONT is its modularity, which allows the activation or deactivation of specific tools, giving great flexibility to the user. Nevertheless, possibly the best feature of CulebrONT is the opportunity to benchmark the selected tool combinations based on the excellent report generated by the pipeline. This HTML report aggregates the output of several tools for quality evaluation of the assemblies (e.g. BUSCO [2] or QUAST [3]) generated by the different assemblers, in addition to the running time and configuration parameters. Such information is of great help to identify the best-suited pipeline, as exemplified by the authors using four datasets of different taxonomic origins. Finally, CulebrONT can handle multiple samples in parallel, which makes it a good solution for laboratories looking for multiple assemblies on a large scale.
References
1. Orjuela J, Comte A, Ravel S, Charriat F, Vi T, Sabot F, Cunnac S (2022) CulebrONT: a streamlined long reads multi-assembler pipeline for prokaryotic and eukaryotic genomes. bioRxiv, 2021.07.19.452922, ver. 5 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2021.07.19.452922
2. Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM (2015) BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics, 31, 3210–3212. https://doi.org/10.1093/bioinformatics/btv351
3. Gurevich A, Saveliev V, Vyahhi N, Tesler G (2013) QUAST: quality assessment tool for genome assemblies. Bioinformatics, 29, 1072–1075. https://doi.org/10.1093/bioinformatics/btt086