A comprehensive look at Xenopus gut microbiota: effects of feed, developmental stages and parental transmission
Recommended by Wirulda Pootakham based on reviews by Vanessa Marcelino and 1 anonymous reviewer
It is well established that the gut microbiota play an important role in the overall health of their hosts (Jandhyala et al. 2015). To date, there are still a limited number of studies on the complex microbial communites inhabiting vertebrate digestive systems, especially the ones that also explored the functional diversity of the microbial community (Bletz et al. 2016).
This preprint by Scalvenzi et al. (2021) reports a comprehensive study on the phylogenetic and metabolic profiles of the Xenopus gut microbiota. The author describes significant changes in the gut microbiome communities at different developmental stages and demonstrates different microbial community composition across organs. In addition, the study also investigates the impact of diet on the Xenopus tadpole gut microbiome communities as well as how the bacterial communities are transmitted from parents to the next generation.
This is one of the first studies that addresses the interactions between gut bacteria and tadpoles during the development. The authors observe the dynamics of gut microbiome communities during tadpole growth and metamorphosis. They also explore host-gut microbial community metabolic interactions and demostrate the capacity of the microbiome to complement the metabolic pathways of the Xenopus genome. Although this study is limited by the use of Xenopus tadpoles in a laboratory, which are probably different from those in nature, I believe it still provides important and valuable information for the research community working on vertebrate’s microbiota and their interaction with the host.
References
Bletz et al. (2016). Amphibian gut microbiota shifts differentially in community structure but converges on habitat-specific predicted functions. Nature Communications, 7(1), 1-12. doi: https://doi.org/10.1038/ncomms13699
Jandhyala, S. M., Talukdar, R., Subramanyam, C., Vuyyuru, H., Sasikala, M., & Reddy, D. N. (2015). Role of the normal gut microbiota. World journal of gastroenterology: WJG, 21(29), 8787. doi: https://dx.doi.org/10.3748%2Fwjg.v21.i29.8787
Scalvenzi, T., Clavereau, I., Bourge, M. & Pollet, N. (2021) Gut microbial ecology of Xenopus tadpoles across life stages. bioRxiv, 2020.05.25.110734, ver. 4 peer-reviewed and recommended by Peer community in Geonmics. https://doi.org/10.1101/2020.05.25.110734
| Gut microbial ecology of Xenopus tadpoles across life stages | Thibault Scalvenzi, Isabelle Clavereau, Mickael Bourge, Nicolas Pollet | <p><strong>Background</strong> The microorganism world living in amphibians is still largely under-represented and under-studied in the literature. Among anuran amphibians, African clawed frogs of the Xenopus genus stand as well-characterized mode... |  | Evolutionary genomics, Metagenomics, Vertebrates | Wirulda Pootakham | | 2020-05-25 14:01:19 | View |
Traces of transposable element in genome dark matter co-opted by flowering gene regulation networks
Agnes Baud, Mariene Wan, Danielle Nouaud, Nicolas Francillonne, Dominique Anxolabehere, Hadi Quesneville
https://doi.org/10.1101/547877
Using small fragments to discover old TE remnants: the Duster approach empowers the TE detection
Recommended by Francois Sabot based on reviews by Josep Casacuberta and 1 anonymous reviewer based on reviews by Josep Casacuberta and 1 anonymous reviewer
Transposable elements are the raw material of the dark matter of the genome, the foundation of the next generation of genes and regulation networks". This sentence could be the essence of the paper of Baud et al. (2021). Transposable elements (TEs) are endogenous mobile genetic elements found in almost all genomes, which were discovered in 1948 by Barbara McClintock (awarded in 1983 the only unshared Medicine Nobel Prize so far). TEs are present everywhere, from a single isolated copy for some elements to more than millions for others, such as Alu. They are founders of major gene lineages (HET-A, TART and telomerases, RAG1/RAG2 proteins from mammals immune system; Diwash et al, 2017), and even of retroviruses (Xiong & Eickbush, 1988). However, most TEs appear as selfish elements that replicate, land in a new genomic region, then start to decay and finally disappear in the midst of the genome, turning into genomic ‘dark matter’ (Vitte et al, 2007). The mutations (single point, deletion, recombination, and so on) that occur during this slow death erase some of their most notable features and signature sequences, rendering them completely unrecognizable after a few million years. Numerous TE detection tools have tried to optimize their detection (Goerner-Potvin & Bourque, 2018), but further improvement is definitely challenging. This is what Baud et al. (2021) accomplished in their paper. They used a simple, elegant and efficient k-mer based approach to find small signatures that, when accumulated, allow identifying very old TEs. Using this method, called Duster, they improved the amount of annotated TEs in the model plant Arabidopsis thaliana by 20%, pushing the part of this genome occupied by TEs up from 40 to almost 50%. They further observed that these very old Duster-specific TEs (i.e., TEs that are only detected by Duster) are, among other properties, close to genes (much more than recent TEs), not targeted by small RNA pathways, and highly associated with conserved regions across the rosid family. In addition, they are highly associated with flowering or stress response genes, and may be involved through exaptation in the evolution of responses to environmental changes. TEs are not just selfish elements: more and more studies have shown their key role in the evolution of their hosts, and tools such as Duster will help us better understand their impact.
References
Baud, A., Wan, M., Nouaud, D., Francillonne, N., Anxolabéhère, D. and Quesneville, H. (2021). Traces of transposable elements in genome dark matter co-opted by flowering gene regulation networks. bioRxiv, 547877, ver. 5 peer-reviewed and recommended by PCI Genomics.doi: https://doi.org/10.1101/547877
Bourque, G., Burns, K.H., Gehring, M. et al. (2018) Ten things you should know about transposable elements. Genome Biology 19:199. doi: https://doi.org/10.1186/s13059-018-1577-z
Goerner-Potvin, P., Bourque, G. Computational tools to unmask transposable elements. Nature Reviews Genetics 19:688–704 (2018) https://doi.org/10.1038/s41576-018-0050-x
Jangam, D., Feschotte, C. and Betrán, E. (2017) Transposable element domestication as an adaptation to evolutionary conflicts. Trends in Genetics 33:817-831. doi: https://doi.org/10.1016/j.tig.2017.07.011
Vitte, C., Panaud, O. and Quesneville, H. (2007) LTR retrotransposons in rice (Oryza sativa, L.): recent burst amplifications followed by rapid DNA loss. BMC Genomics 8:218. doi: https://doi.org/10.1186/1471-2164-8-218
Xiong, Y. and Eickbush, T. H. (1988) Similarity of reverse transcriptase-like sequences of viruses, transposable elements, and mitochondrial introns. Molecular Biology and Evolution 5: 675–690. doi: https://doi.org/10.1093/oxfordjournals.molbev.a040521
| Traces of transposable element in genome dark matter co-opted by flowering gene regulation networks | Agnes Baud, Mariene Wan, Danielle Nouaud, Nicolas Francillonne, Dominique Anxolabehere, Hadi Quesneville | <p>Transposable elements (TEs) are mobile, repetitive DNA sequences that make the largest contribution to genome bulk. They thus contribute to the so-called 'dark matter of the genome', the part of the genome in which nothing is immediately recogn... |  | Bioinformatics, Evolutionary genomics, Functional genomics, Plants, Structural genomics, Viruses and transposable elements | Francois Sabot | Anonymous, Josep Casacuberta | 2020-04-07 17:12:12 | View |
An evaluation of pool-sequencing transcriptome-based exon capture for population genomics in non-model species
Emeline Deleury, Thomas Guillemaud, Aurélie Blin & Eric Lombaert
https://doi.org/10.1101/583534
Assessing a novel sequencing-based approach for population genomics in non-model species
Recommended by Thomas Derrien and Sebastian Ernesto Ramos-Onsins based on reviews by Valentin Wucher and 1 anonymous reviewer
Developing new sequencing and bioinformatic strategies for non-model species is of great interest in many applications, such as phylogenetic studies of diverse related species, but also for studies in population genomics, where a relatively large number of individuals is necessary. Different approaches have been developed and used in these last two decades, such as RAD-Seq (e.g., Miller et al. 2007), exome sequencing (e.g., Teer and Mullikin 2010) and other genome reduced representation methods that avoid the use of a good reference and well annotated genome (reviewed at Davey et al. 2011). However, population genomics studies require the analysis of numerous individuals, which makes the studies still expensive. Pooling samples was thought as an inexpensive strategy to obtain estimates of variability and other related to the frequency spectrum, thus allowing the study of variability at population level (e.g., Van Tassell et al. 2008), although the major drawback was the loss of information related to the linkage of the variants. In addition, population analysis using all these sequencing strategies require statistical and empirical validations that are not always fully performed. A number of studies aiming to obtain unbiased estimates of variability using reduced representation libraries and/or with pooled data have been performed (e.g., Futschik and Schlötterer 2010, Gautier et al. 2013, Ferretti et al. 2013, Lynch et al. 2014), as well as validation of new sequencing methods for population genetic analyses (e.g., Gautier et al. 2013, Nevado et al. 2014). Nevertheless, empirical validation using both pooled and individual experimental approaches combined with different bioinformatic methods has not been always performed.
Here, Deleury et al. (2020) proposed an efficient and elegant way of quantifying the single-nucleotide polymorphisms (SNPs) of exon-derived sequences in a non-model species (i.e. for which no reference genome sequence is available) at the population level scale. They also designed a new procedure to capture exon-derived sequences based on a reference transcriptome. In addition, they were able to make predictions of intron-exon boundaries for de novo transcripts based on the decay of read depth at the ends of the coding regions.
Based on theoretical predictions (Gautier et al. 2013), Deleury et al. (2020) designed a procedure to test the accuracy of variant allele frequencies (AFs) with pooled samples, in a reduced genome-sequence library made with transcriptome regions, and additionally testing the effects of new bioinformatic methods in contrast to standardized methods. They applied their strategy on the non-model species Asian ladybird (Harmonia axyridis), for which a draft genome is available, thereby allowing them to benchmark their method with regard to a traditional mapping-based approach. Based on species-specific de novo transcriptomes, they designed capture probes which are then used to call SNPx and then compared the resulting SNP AFs at the individual (multiplexed) versus population (pooled) levels. Interestingly, they showed that SNP AFs in the pool sequencing strategy nicely correlate with the individual ones but obviously in a cost-effective way. Studies of population genomics for non-model species have usually limited budgets. The number of individuals required for population genomics analysis multiply the costs of the project, making pooling samples an interesting option. Furthermore, the use of pool sequencing is not always a choice, as many organisms are too small and/or individuals are too sticked each other to be individually sequenced (e.g., Choquet et al. 2019, Kurland et al. 2019). In addition, the study of a reduced section of the genome is cheaper and often sufficient for a number of population genetic questions, such as the understanding of general demographic events, or the estimation of the effects of positive and/or negative selection at functional coding regions. Studies on population genomics of non-model species have many applications in related fields, such as conservation genetics, control of invasive species, etc. The work of Deleury et al. (2020) is an elegant contribution to the assessment and validation of new methodologies used for the analysis of genome variations at the intra-population variability level, highlighting straight bioinformatic and reliable sequencing methods for population genomics studies.
References
[1] Choquet et al. (2019). Towards population genomics in non-model species with large genomes: a case study of the marine zooplankton Calanus finmarchicus. Royal Society open science, 6(2), 180608. doi: https://doi.org/10.1098/rsos.180608
[2] Davey, J. W., Hohenlohe, P. A., Etter, P. D., Boone, J. Q., Catchen, J. M. and Blaxter, M. L. (2011). Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nature Reviews Genetics, 12(7), 499-510. doi: https://doi.org/10.1038/nrg3012
[3] Deleury, E., Guillemaud, T., Blin, A. and Lombaert, E. (2020) An evaluation of pool-sequencing transcriptome-based exon capture for population genomics in non-model species. bioRxiv, 10.1101/583534, ver. 7 peer-reviewed and recommended by PCI Genomics. https://doi.org/10.1101/583534
[4] Ferretti, L., Ramos‐Onsins, S. E. and Pérez‐Enciso, M. (2013). Population genomics from pool sequencing. Molecular ecology, 22(22), 5561-5576. doi: https://doi.org/10.1111/mec.12522
[5] Futschik, A. and Schlötterer, C. (2010). Massively parallel sequencing of pooled DNA samples—the next generation of molecular markers. Genetics, 186 (1), 207-218. doi: https://doi.org/10.1534/genetics.110.114397
[6] Gautier et al. (2013). Estimation of population allele frequencies from next‐generation sequencing data: pool‐versus individual‐based genotyping. Molecular Ecology, 22(14), 3766-3779. doi: https://doi.org/10.1111/mec.12360
[7] Kurland et al. (2019). Exploring a Pool‐seq‐only approach for gaining population genomic insights in nonmodel species. Ecology and evolution, 9(19), 11448-11463. doi: https://doi.org/10.1002/ece3.5646
[8] Lynch, M., Bost, D., Wilson, S., Maruki, T. and Harrison, S. (2014). Population-genetic inference from pooled-sequencing data. Genome biology and evolution, 6(5), 1210-1218. doi: https://doi.org/10.1093/gbe/evu085
[9] Miller, M. R., Dunham, J. P., Amores, A., Cresko, W. A. and Johnson, E. A. (2007). Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers. Genome research, 17(2), 240-248. doi: https://doi.org/10.1101%2Fgr.5681207
[10] Nevado, B., Ramos‐Onsins, S. E. and Perez‐Enciso, M. (2014). Resequencing studies of nonmodel organisms using closely related reference genomes: optimal experimental designs and bioinformatics approaches for population genomics. Molecular ecology, 23(7), 1764-1779. doi: https://doi.org/10.1111/mec.12693
[11] Teer, J. K. and Mullikin, J. C. (2010). Exome sequencing: the sweet spot before whole genomes. Human molecular genetics, 19(R2), R145-R151. doi: https://doi.org/10.1093/hmg/ddq333
[12] Van Tassell et al. (2008). SNP discovery and allele frequency estimation by deep sequencing of reduced representation libraries. Nature methods, 5(3), 247-252. doi: https://doi.org/10.1038/nmeth.1185
| An evaluation of pool-sequencing transcriptome-based exon capture for population genomics in non-model species | Emeline Deleury, Thomas Guillemaud, Aurélie Blin & Eric Lombaert | <p>Exon capture coupled to high-throughput sequencing constitutes a cost-effective technical solution for addressing specific questions in evolutionary biology by focusing on expressed regions of the genome preferentially targeted by selection. Tr... | 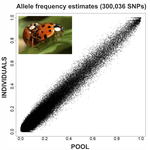 | Bioinformatics, Population genomics | Thomas Derrien | | 2020-02-26 09:21:11 | View |
A rapid and simple method for assessing and representing genome sequence relatedness
M Briand, M Bouzid, G Hunault, M Legeay, M Fischer-Le Saux, M Barret
https://doi.org/10.1101/569640
A quick alternative method for resolving bacterial taxonomy using short identical DNA sequences in genomes or metagenomes
Recommended by B. Jesse Shapiro based on reviews by Gavin Douglas and 1 anonymous reviewer
The bacterial species problem can be summarized as follows: bacteria recombine too little, and yet too much (Shapiro 2019).
Too little in the sense that recombination is not obligately coupled with reproduction, as in sexual eukaryotes. So the Biological Species Concept (BSC) of reproductive isolation does not strictly apply to clonally reproducing organisms like bacteria. Too much in the sense that genetic exchange can occur promiscuously across species (or even Domains), potentially obscuring species boundaries.
In parallel to such theoretical considerations, several research groups have taken more pragmatic approaches to defining bacterial species based on sequence similarity cutoffs, such as genome-wide average nucleotide identity (ANI). At a cutoff above 95% ANI, genomes are considered to come from the same species. While this cutoff may appear arbitrary, a discontinuity around 95% in the distribution of ANI values has been argued to provide a 'natural' cutoff (Jain et al. 2018). This discontinuity has been criticized as being an artefact of various biases in genome databases (Murray, Gao, and Wu 2020), but appears to be a general feature of relatively unbiased metagenome-assembled genomes as well (Olm et al. 2020). The 95% cutoff has been suggested to represent a barrier to homologous recombination (Olm et al. 2020), although clusters of genetic exchange consistent with BSC-like species are observed at much finer identity cutoffs (Shapiro 2019; Arevalo et al. 2019).
Although 95% ANI is the most widely used genomic standard for species delimitation, it is by no means the only plausible approach. In particular, tracts of identical DNA provide evidence for recent genetic exchange, which in turn helps define BSC-like clusters of genomes (Arevalo et al. 2019). In this spirit, Briand et al. (2020) introduce a genome-clustering method based on the number of shared identical DNA sequences of length k (or k-mers). Using a test dataset of Pseudomonas genomes, they find that 95% ANI corresponds to approximately 50% of shared 15-mers. Applying this cutoff yields 350 Pseudomonas species, whereas the current taxonomy only includes 207 recognized species. To determine whether splitting the genus into a greater number of species is at all useful, they compare their new classification scheme to the traditional one in terms of the ability to taxonomically classify metagenomic sequencing reads from three Pseudomonas-rich environments. In all cases, the new scheme (termed K-IS for "Kinship relationships Identification with Shared k-mers") yielded a higher number of classified reads, with an average improvement of 1.4-fold. This is important because increasing the number of genome sequences in a reference database – without consistent taxonomic annotation of these genomes – paradoxically leads to fewer classified metagenomic reads. Thus a rapid, automated taxonomy such as the one proposed here offers an opportunity to more fully harness the information from metagenomes.
KI-S is also fast to run, so it is feasible to test several values of k and quickly visualize the clustering using an interactive, zoomable circle-packing display (that resembles a cross-section of densely packed, three-dimensional dendrogram). This interface allows the rapid flagging of misidentified species, or understudied species with few sequenced representatives as targets for future study. Hopefully these initial Pseudomonas results will inspire future studies to apply the method to additional taxa, and to further characterize the relationship between ANI and shared identical k-mers. Ultimately, I hope that such investigations will resolve the issue of whether or not there is a 'natural' discontinuity for bacterial species, and what evolutionary forces maintain this cutoff.
References
Arevalo P, VanInsberghe D, Elsherbini J, Gore J, Polz MF (2019) A Reverse Ecology Approach Based on a Biological Definition of Microbial Populations. Cell, 178, 820-834.e14. https://doi.org/10.1016/j.cell.2019.06.033
Briand M, Bouzid M, Hunault G, Legeay M, Saux MF-L, Barret M (2020) A rapid and simple method for assessing and representing genome sequence relatedness. bioRxiv, 569640, ver. 5 peer-reveiwed and recommended by PCI Genomics. https://doi.org/10.1101/569640
Jain C, Rodriguez-R LM, Phillippy AM, Konstantinidis KT, Aluru S (2018) High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nature Communications, 9, 5114. https://doi.org/10.1038/s41467-018-07641-9
Murray CS, Gao Y, Wu M (2020) There is no evidence of a universal genetic boundary among microbial species. bioRxiv, 2020.07.27.223511. https://doi.org/10.1101/2020.07.27.223511
Olm MR, Crits-Christoph A, Diamond S, Lavy A, Carnevali PBM, Banfield JF (2020) Consistent Metagenome-Derived Metrics Verify and Delineate Bacterial Species Boundaries. mSystems, 5. https://doi.org/10.1128/mSystems.00731-19
Shapiro BJ (2019) What Microbial Population Genomics Has Taught Us About Speciation. In: Population Genomics: Microorganisms Population Genomics. (eds Polz MF, Rajora OP), pp. 31–47. Springer International Publishing, Cham. https://doi.org/10.1007/13836201810
| A rapid and simple method for assessing and representing genome sequence relatedness | M Briand, M Bouzid, G Hunault, M Legeay, M Fischer-Le Saux, M Barret | <p>Coherent genomic groups are frequently used as a proxy for bacterial species delineation through computation of overall genome relatedness indices (OGRI). Average nucleotide identity (ANI) is a widely employed method for estimating relatedness ... |  | Bioinformatics, Metagenomics | B. Jesse Shapiro | Gavin Douglas | 2019-11-07 16:37:56 | View |





