
Latest recommendations

| Id | Title * | Authors * | Abstract * ▲ | Picture * | Thematic fields * | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
16 Dec 2022
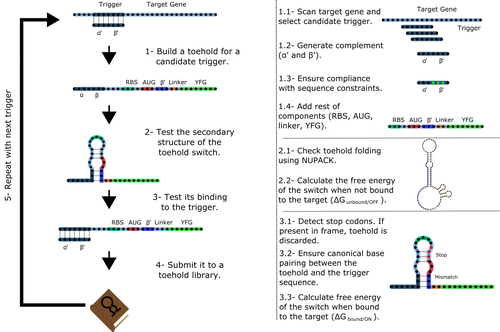
Toeholder: a Software for Automated Design and In Silico Validation of Toehold RiboswitchesA novel approach for engineering biological systems by interfacing computer science with synthetic biologyRecommended by Sahar Melamed based on reviews by Wim Wranken and 1 anonymous reviewerBiological systems depend on finely tuned interactions of their components. Thus, regulating these components is critical for the system's functionality. In prokaryotic cells, riboswitches are regulatory elements controlling transcription or translation. Riboswitches are RNA molecules that are usually located in the 5′-untranslated region of protein-coding genes. They generate secondary structures leading to the regulation of the expression of the downstream protein-coding gene (Kavita and Breaker, 2022). Riboswitches are very versatile and can bind a wide range of small molecules; in many cases, these are metabolic byproducts from the gene’s enzymatic or signaling pathway. Their versatility and abundance in many species make them attractive for synthetic biological circuits. One class that has been drawing the attention of synthetic biologists is toehold switches (Ekdahl et al., 2022; Green et al., 2014). These are single-stranded RNA molecules harboring the necessary elements for translation initiation of the downstream gene: a ribosome-binding site and a start codon. Conformation change of toehold switches is triggered by an RNA molecule, which enables translation. To exploit the most out of toehold switches, automation of their design would be highly advantageous. Cisneros and colleagues (Cisneros et al., 2022) developed a tool, “Toeholder”, that automates the design of toehold switches and performs in silico tests to select switch candidates for a target gene. Toeholder is an open-source tool that provides a comprehensive and automated workflow for the design of toehold switches. While web tools have been developed for designing toehold switches (To et al., 2018), Toeholder represents an intriguing approach to engineering biological systems by coupling synthetic biology with computational biology. Using molecular dynamics simulations, it identified the positions in the toehold switch where hydrogen bonds fluctuate the most. Identifying these regions holds great potential for modifications when refining the design of the riboswitches. To be effective, toehold switches should provide a strong ON signal and a weak OFF signal in the presence or the absence of a target, respectively. Toeholder nicely ranks the candidate toehold switches based on experimental evidence that correlates with toehold performance (based on good ON/OFF ratios). Riboswitches are highly appealing for a broad range of applications, including pharmaceutical and medical purposes (Blount and Breaker, 2006; Giarimoglou et al., 2022; Tickner and Farzan, 2021), thanks to their adaptability and inexpensiveness. The Toeholder tool developed by Cisneros and colleagues is expected to promote the implementation of toehold switches into these various applications. References Blount KF, Breaker RR (2006) Riboswitches as antibacterial drug targets. Nature Biotechnology, 24, 1558–1564. https://doi.org/10.1038/nbt1268 Cisneros AF, Rouleau FD, Bautista C, Lemieux P, Dumont-Leblond N, ULaval 2019 T iGEM (2022) Toeholder: a Software for Automated Design and In Silico Validation of Toehold Riboswitches. bioRxiv, 2021.11.09.467922, ver. 3 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2021.11.09.467922 Ekdahl AM, Rojano-Nisimura AM, Contreras LM (2022) Engineering Toehold-Mediated Switches for Native RNA Detection and Regulation in Bacteria. Journal of Molecular Biology, 434, 167689. https://doi.org/10.1016/j.jmb.2022.167689 Giarimoglou N, Kouvela A, Maniatis A, Papakyriakou A, Zhang J, Stamatopoulou V, Stathopoulos C (2022) A Riboswitch-Driven Era of New Antibacterials. Antibiotics, 11, 1243. https://doi.org/10.3390/antibiotics11091243 Green AA, Silver PA, Collins JJ, Yin P (2014) Toehold Switches: De-Novo-Designed Regulators of Gene Expression. Cell, 159, 925–939. https://doi.org/10.1016/j.cell.2014.10.002 Kavita K, Breaker RR (2022) Discovering riboswitches: the past and the future. Trends in Biochemical Sciences. https://doi.org/10.1016/j.tibs.2022.08.009 Tickner ZJ, Farzan M (2021) Riboswitches for Controlled Expression of Therapeutic Transgenes Delivered by Adeno-Associated Viral Vectors. Pharmaceuticals, 14, 554. https://doi.org/10.3390/ph14060554 To AC-Y, Chu DH-T, Wang AR, Li FC-Y, Chiu AW-O, Gao DY, Choi CHJ, Kong S-K, Chan T-F, Chan K-M, Yip KY (2018) A comprehensive web tool for toehold switch design. Bioinformatics, 34, 2862–2864. https://doi.org/10.1093/bioinformatics/bty216 | Toeholder: a Software for Automated Design and In Silico Validation of Toehold Riboswitches | Angel F. Cisneros, François D. Rouleau, Carla Bautista, Pascale Lemieux, Nathan Dumont-Leblond | <p>Abstract: Synthetic biology aims to engineer biological circuits, which often involve gene expression. A particularly promising group of regulatory elements are riboswitches because of their versatility with respect to their targets, but e... | | Bioinformatics | Sahar Melamed | 2022-02-16 14:40:13 | ||
15 Mar 2024
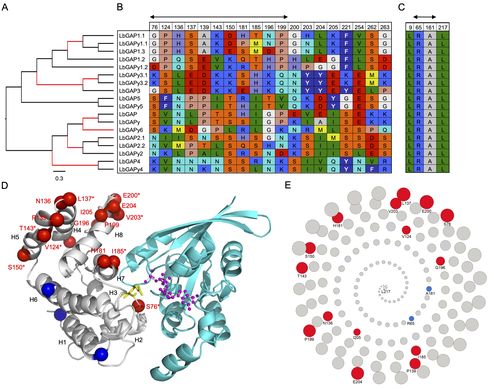
Convergent origin and accelerated evolution of vesicle-associated RhoGAP proteins in two unrelated parasitoid waspsUsing transcriptomics and proteomics to understand the expansion of a secreted poisonous armoury in parasitoid wasps genomesRecommended by Ignacio Bravo based on reviews by Inacio Azevedo and 2 anonymous reviewers based on reviews by Inacio Azevedo and 2 anonymous reviewers
Parasitoid wasps lay their eggs inside another arthropod, whose body is physically consumed by the parasitoid larvae. Phylogenetic inference suggests that Parasitoida are monophyletic, and that this clade underwent a strong radiation shortly after branching off from the Apocrita stem, some 236 million years ago (Peters et al. 2017). The increase in taxonomic diversity during evolutionary radiations is usually concurrent with an increase in genetic/genomic diversity, and is often associated with an increase in phenotypic diversity. Gene (or genome) duplication provides the evolutionary potential for such increase of genomic diversity by neo/subfunctionalisation of one of the gene paralogs, and is often proposed to be related to evolutionary radiations (Ohno 1970; Francino 2005).
References
| Convergent origin and accelerated evolution of vesicle-associated RhoGAP proteins in two unrelated parasitoid wasps | Dominique Colinet, Fanny Cavigliasso, Matthieu Leobold, Appoline Pichon, Serge Urbach, Dominique Cazes, Marine Poullet, Maya Belghazi, Anne-Nathalie Volkoff, Jean-Michel Drezen, Jean-Luc Gatti, and Marylène Poirié | <p>Animal venoms and other protein-based secretions that perform a variety of functions, from predation to defense, are highly complex cocktails of bioactive compounds. Gene duplication, accompanied by modification of the expression and/or functio... | | Evolutionary genomics | Ignacio Bravo | 2023-06-12 11:08:31 | ||
03 Jul 2024
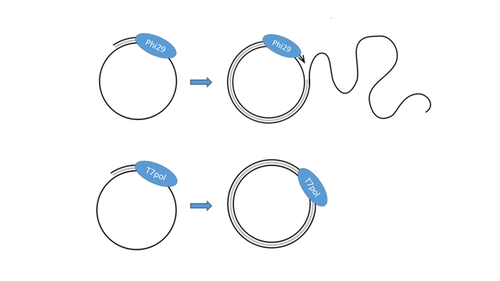
T7 DNA polymerase treatment improves quantitative sequencing of both double-stranded and single-stranded DNA virusesImproving the sequencing of single-stranded DNA viruses: Another brick for building Earth's complete virome encyclopediaRecommended by Sebastien Massart based on reviews by Philippe Roumagnac and 3 anonymous reviewersThe wide adoption of high-throughput sequencing technologies has uncovered an astonishing diversity of viruses in most biosphere habitats. Among them, single-stranded DNA viruses are prevalent, infecting diverse hosts from all three domains of life (Malathi et al. 2014) with some species being highly pathogenic to animals or plants. Sequencing of single-stranded DNA viruses requires a specific approach that usually leads to their over-representation compared to double-stranded DNA. The article from Billaud et al. (2024) addresses this challenge. It presents a novel and efficient method for converting single-stranded DNA to double-stranded DNA using T7 DNA polymerase before high-throughput virome sequencing. It compares this new method with the Phi29 polymerase method, demonstrating its advantages in the representation and accuracy of viral DNA content in well-defined synthetic phage mixtures and complex human virome samples from the stool. This T7 DNA polymerase treatment significantly improved the richness and abundance of the Microviridae fraction in their samples, suggesting a more comprehensive representation of viral diversity. The article presents a compelling case for testing and adopting the T7 DNA polymerase methodology in preparing virome samples for shotgun sequencing. This novel approach, supported by comparative analysis with existing methodologies, represents a valuable contribution to metagenomics for characterizing virome diversity.
References Billaud M, Theodorou I, Lamy-Besnier Q, Shah SA, Lecointe F, Sordi LD, Paepe MD, Petit M-A (2024) T7 DNA polymerase treatment improves quantitative sequencing of both double-stranded and single-stranded DNA viruses. bioRxiv, ver. 4 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2022.12.12.520144 Malathi VG, Renuka Devi P. (2019) ssDNA viruses: key players in global virome. Virus disease. 30: 3–12. https://doi.org/10.1007/s13337-019-00519-4
| T7 DNA polymerase treatment improves quantitative sequencing of both double-stranded and single-stranded DNA viruses | Maud Billaud, Ilias Theodorou, Quentin Lamy-Besnier, Shiraz Shah, François Lecointe, Luisa De Sordi, Marianne De Paepe, Marie-Agnès Petit | <p>Background: Bulk microbiome, as well as virome-enriched shotgun sequencing only reveals the double-stranded DNA (dsDNA) content of a given sample, unless specific treatments are applied. However, genomes of viruses often consist of a circular s... | | Viruses and transposable elements | Sebastien Massart | 2023-12-20 16:50:00 | ||
06 Apr 2021
Evidence for shared ancestry between Actinobacteria and Firmicutes bacteriophagesViruses of bacteria: phages evolution across phylum boundariesRecommended by Denis Tagu based on reviews by 3 anonymous reviewersBacteria and phages have coexisted and coevolved for a long time. Phages are bacteria-infecting viruses, with a symbiotic status sensu lato, meaning they can be pathogenic, commensal or mutualistic. Thus, the association between bacteria phages has probably played a key role in the high adaptability of bacteria to most - if not all – of Earth’s ecosystems, including other living organisms (such as eukaryotes), and also regulate bacterial community size (for instance during bacterial blooms). As genetic entities, phages are submitted to mutations and natural selection, which changes their DNA sequence. Therefore, comparative genomic analyses of contemporary phages can be useful to understand their evolutionary dynamics. International initiatives such as SEA-PHAGES have started to tackle the issue of history of phage-bacteria interactions and to describe the dynamics of the co-evolution between bacterial hosts and their associated viruses. Indeed, the understanding of this cross-talk has many potential implications in terms of health and agriculture, among others. The work of Koert et al. (2021) deals with one of the largest groups of bacteria (Actinobacteria), which are Gram-positive bacteria mainly found in soil and water. Some soil-born Actinobacteria develop filamentous structures reminiscent of the mycelium of eukaryotic fungi. In this study, the authors focused on the Streptomyces clade, a large genus of Actinobacteria colonized by phages known for their high level of genetic diversity. The authors tested the hypothesis that large exchanges of genetic material occurred between Streptomyces and diverse phages associated with bacterial hosts. Using public datasets, their comparative phylogenomic analyses identified a new cluster among Actinobacteria–infecting phages closely related to phages of Firmicutes. Moreover, the GC content and codon-usage biases of this group of phages of Actinobacteria are similar to those of Firmicutes. This work demonstrates for the first time the transfer of a bacteriophage lineage from one bacterial phylum to another one. The results presented here suggest that the age of the described transfer is probably recent since several genomic characteristics of the phage are not fully adapted to their new hosts. However, the frequency of such transfer events remains an open question. If frequent, such exchanges would mean that pools of bacteriophages are regularly fueled by genetic material coming from external sources, which would have important implications for the co-evolutionary dynamics of phages and bacteria. References Koert, M., López-Pérez, J., Courtney Mattson, C., Caruso, S. and Erill, I. (2021) Evidence for shared ancestry between Actinobacteria and Firmicutes bacteriophages. bioRxiv, 842583, version 5 peer-reviewed and recommended by Peer community in Genomics. doi: https://doi.org/10.1101/842583 | Evidence for shared ancestry between Actinobacteria and Firmicutes bacteriophages | Matthew Koert, Júlia López-Pérez, Courtney Mattson, Steven M. Caruso, Ivan Erill | <p>Bacteriophages typically infect a small set of related bacterial strains. The transfer of bacteriophages between more distant clades of bacteria has often been postulated, but remains mostly unaddressed. In this work we leverage the sequencing ... | | Evolutionary genomics | Denis Tagu | 2019-12-10 15:26:31 | ||
06 Feb 2024

The need of decoding life for taking care of biodiversity and the sustainable use of nature in the Anthropocene - a Faroese perspectiveWhy sequence everything? A raison d’être for the Genome Atlas of Faroese EcologyRecommended by Stephen Richards based on reviews by Tereza Manousaki and 1 anonymous reviewer
When discussing the Earth BioGenome Project with scientists and potential funding agencies, one common question is: why sequence everything? Whether sequencing a subset would be more optimal is not an unreasonable question given what we know about the mathematics of importance and Pareto’s 80:20 principle, that 80% of the benefits can come from 20% of the effort. However, one must remember that this principle is an observation made in hindsight and selecting the most effective 20% of experiments is difficult. As an example, few saw great applied value in comparative genomic analysis of the archaea Haloferax mediterranei, but this enabled the discovery of CRISPR/Cas9 technology (1). When discussing whether or not to sequence all life on our planet, smaller countries such as the Faroe Islands are seldom mentioned.
1 Mojica, F. J., Díez-Villaseñor, C. S., García-Martínez, J. & Soria, E. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J Mol Evol 60, 174-182 (2005). 2 Mikalsen, S-O., Hjøllum, J. í., Salter, I., Djurhuus, A. & Kongsstovu, S. í. The need of decoding life for taking care of biodiversity and the sustainable use of nature in the Anthropocene – a Faroese perspective. EcoEvoRxiv (2024), ver. 3 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.32942/X21S4C | The need of decoding life for taking care of biodiversity and the sustainable use of nature in the Anthropocene - a Faroese perspective | Svein-Ole Mikalsen, Jari í Hjøllum, Ian Salter, Anni Djurhuus, Sunnvør í Kongsstovu | <p>Biodiversity is under pressure, mainly due to human activities and climate change. At the international policy level, it is now recognised that genetic diversity is an important part of biodiversity. The availability of high-quality reference g... | | ERGA, ERGA Pilot, Population genomics, Vertebrates | Stephen Richards | 2023-07-31 16:59:33 | ||
01 May 2024
Evolution of ion channels in cetaceans: A natural experiment in the tree of lifePositive selection acted upon cetacean ion channels during the aquatic transitionRecommended by Gavin Douglas based on reviews by 2 anonymous reviewers
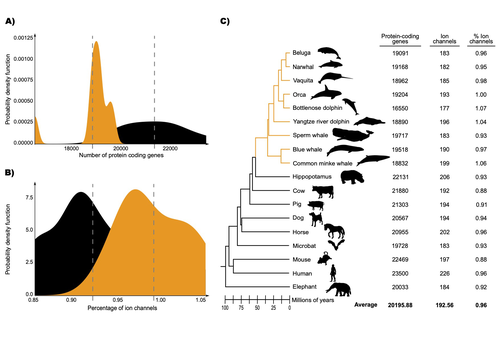
The transition of cetaceans (whales, dolphins, and porpoises) from terrestrial to aquatic lifestyles is a striking example of natural selection driving major phenotypic changes (Figure 1). For instance, cetaceans have evolved the ability to withstand high pressure and to store oxygen for long periods, among other adaptations (Das et al. 2023). Many phenotypic changes, such as shifts in organ structure, have been well-characterized through fossils (Thewissen et al. 2009). Although such phenotypic transitions are now well understood, we have only a partial understanding of the underlying genetic mechanisms. Scanning for signatures of adaptation in genes related to phenotypes of interest is one approach to better understand these mechanisms. This was the focus of Uribe and colleagues’ (2024) work, who tested for such signatures across cetacean protein-coding genes.
Figure 1: The skeletons of Ambulocetus (an early whale; top) and Pakicetus (the earliest known cetacean, which lived about 50 million years ago; bottom). Copyright: J. G. M. Thewissen. Displayed here with permission from the copyright holder.
The authors were specifically interested in investigating the evolution of ion channels, as these proteins play fundamental roles in physiological processes. An important aspect of their work was to develop a bioinformatic pipeline to identify orthologous ion channel genes across a set of genomes. After applying their bioinformatic workflow to 18 mammalian species (including nine cetaceans), they conducted tests to find out whether these genes showed signatures of positive selection in the cetacean lineage. For many ion channel genes, elevated ratios of non-synonymous to synonymous substitution rates were detected (for at least a subset of sites, and not necessarily the entire coding region of the genes). The genes concerned were enriched for several functions, including heart and nervous system-related phenotypes. One top gene hit among the putatively selected genes was SCN5A, which encodes a sodium channel expressed in the heart. Interestingly, the authors noted a specific amino acid replacement, which is associated with sensitivity to the toxin tetrodotoxin in other lineages. This substitution appears to have occurred in the common ancestor of toothed whales, and then was reversed in the ancestor of bottlenose dolphins. The authors describe known bottlenose dolphin interactions with toxin-producing pufferfish that could result in high tetrodotoxin exposure, and thus perhaps higher selection for tetrodotoxin resistance. Although this observation is intriguing, the authors emphasize it requires experimental confirmation. The authors also recapitulated the previously described observation (Yim et al. 2014; Huelsmann et al. 2019) that cetaceans have fewer protein-coding genes compared to terrestrial mammals, on average. This signal has previously been hypothesized to partially reflect adaptive gene loss. For example, specific gene loss events likely decreased the risk of developing blood clots while diving (Huelsmann et al. 2019). Uribe and colleagues also considered overall gene turnover rate, which encompasses gene copy number variation across lineages, and found the cetacean gene turnover rate to be three times higher than that of terrestrial mammals. Finally, they found that cetaceans have a higher proportion of ion channel genes (relative to all protein-coding genes in a genome) compared to terrestrial mammals. Similar investigations of the relative non-synonymous to synonymous substitution rates across cetacean and terrestrial mammal orthologs have been conducted previously, but these have primarily focused on dolphins as the sole cetacean representative (McGowen et al. 2012; Nery et al. 2013; Sun et al. 2013). These projects have also been conducted across a large proportion of orthologous genes, rather than a subset with a particular function. Performing proteome-wide investigations can be valuable in that they summarize the genome-wide signal, but can suffer from a high multiple testing burden. More generally, investigating a more targeted question, such as the extent of positive selection acting on ion channels in this case, or on genes potentially linked to cetaceans’ increased brain sizes (McGowen et al. 2011) or hypoxia tolerance (Tian et al. 2016), can be easier to interpret, as opposed to summarizing broader signals. However, these smaller-scale studies can also experience a high multiple testing burden, especially as similar tests are conducted across numerous studies, which often is not accounted for (Ioannidis 2005). In addition, integrating signals across the entire genome will ultimately be needed given that many genetic changes undoubtedly underlie cetaceans’ phenotypic diversification. As highlighted by the fact that past genome-wide analyses have produced some differing biological interpretations (McGowen et al. 2012; Nery et al. 2013; Sun et al. 2013), this is not a trivial undertaking. Nonetheless, the work performed in this preprint, and in related research, is valuable for (at least) three reasons. First, although it is a challenging task, a better understanding of the genetic basis of cetacean phenotypes could have benefits for many aspects of cetacean biology, including conservation efforts. In addition, the remarkable phenotypic shifts in cetaceans make the question of what genetic mechanisms underlie these changes intrinsically interesting to a wide audience. Last, since the cetacean fossil record is especially well-documented (Thewissen et al. 2009), cetaceans represent an appealing system to validate and further develop statistical methods for inferring adaptation from genetic data. Uribe and colleagues’ (2024) analyses provide useful insights relevant to each of these points, and have generated intriguing hypotheses for further investigation.
Das K, Sköld H, Lorenz A, Parmentier E (2023) Who are the marine mammals? In: “Marine Mammals: A Deep Dive into the World of Science”. Brennecke D, Knickmeier K, Pawliczka I, Siebert U, Wahlberg, M (editors). Springer, Cham. p. 1–14. https://doi.org/10.1007/978-3-031-06836-2_1 Huelsmann M, Hecker N, Springer MS, Gatesy J, Sharma V, Hiller M (2019) Genes lost during the transition from land to water in cetaceans highlight genomic changes associated with aquatic adaptations. Science Advances, 5, eaaw6671. https://doi.org/10.1126/sciadv.aaw6671 Ioannidis JPA (2005) Why most published research findings are false. PLOS Medicine, 2, e124. https://doi.org/10.1371/journal.pmed.0020124 McGowen MR, Montgomery SH, Clark C, Gatesy J (2011) Phylogeny and adaptive evolution of the brain-development gene microcephalin (MCPH1) in cetaceans. BMC Evolutionary Biology, 11, 98. https://doi.org/10.1186/1471-2148-11-98 McGowen MR, Grossman LI, Wildman DE (2012) Dolphin genome provides evidence for adaptive evolution of nervous system genes and a molecular rate slowdown. Proceedings of the Royal Society B: Biological Sciences, 279, 3643–3651. https://doi.org/10.1098/rspb.2012.0869 Nery MF, González DJ, Opazo JC (2013) How to make a dolphin: molecular signature of positive selection in cetacean genome. PLOS ONE, 8, e65491. https://doi.org/10.1371/journal.pone.0065491 Sun YB, Zhou WP, Liu HQ, Irwin DM, Shen YY, Zhang YP (2013) Genome-wide scans for candidate genes involved in the aquatic adaptation of dolphins. Genome Biology and Evolution, 5, 130–139. https://doi.org/10.1093/gbe/evs123 Tian R, Wang Z, Niu X, Zhou K, Xu S, Yang G (2016) Evolutionary genetics of hypoxia tolerance in cetaceans during diving. Genome Biology and Evolution, 8, 827–839. https://doi.org/10.1093/gbe/evw037 Thewissen JGM, Cooper LN, George JC, Bajpai (2009) From land to water: the origin of whales, dolphins, and porpoises. Evolution: Education and Outreach, 2, 272–288. https://doi.org/10.1007/s12052-009-0135-2 Uribe C, Nery M, Zavala K, Mardones G, Riadi G, Opazo J (2024) Evolution of ion channels in cetaceans: A natural experiment in the tree of life. bioRxiv, ver. 8 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2023.06.15.545160 Yim HS, Cho YS, Guang X, Kang SG, Jeong JY, Cha SS, Oh HM, Lee JH, Yang EC, Kwon KK, et al. (2014) Minke whale genome and aquatic adaptation in cetaceans. Nature Genetics, 46, 88–92. https://doi.org/10.1038/ng.2835
| Evolution of ion channels in cetaceans: A natural experiment in the tree of life | Cristóbal Uribe, Mariana F. Nery, Kattina Zavala, Gonzalo A. Mardones, Gonzalo Riadi & Juan C. Opazo | <p>Cetaceans could be seen as a natural experiment within the tree of life in which a mammalian lineage changed from terrestrial to aquatic habitats. This shift involved extensive phenotypic modifications, which represent an opportunity to explore... | | Evolutionary genomics | Gavin Douglas | 2023-07-04 20:53:46 | ||
24 Sep 2020

A rapid and simple method for assessing and representing genome sequence relatednessA quick alternative method for resolving bacterial taxonomy using short identical DNA sequences in genomes or metagenomesRecommended by B. Jesse Shapiro based on reviews by Gavin Douglas and 1 anonymous reviewerThe bacterial species problem can be summarized as follows: bacteria recombine too little, and yet too much (Shapiro 2019). References Arevalo P, VanInsberghe D, Elsherbini J, Gore J, Polz MF (2019) A Reverse Ecology Approach Based on a Biological Definition of Microbial Populations. Cell, 178, 820-834.e14. https://doi.org/10.1016/j.cell.2019.06.033 | A rapid and simple method for assessing and representing genome sequence relatedness | M Briand, M Bouzid, G Hunault, M Legeay, M Fischer-Le Saux, M Barret | <p>Coherent genomic groups are frequently used as a proxy for bacterial species delineation through computation of overall genome relatedness indices (OGRI). Average nucleotide identity (ANI) is a widely employed method for estimating relatedness ... | | Bioinformatics, Metagenomics | B. Jesse Shapiro | Gavin Douglas | 2019-11-07 16:37:56 | |
23 Aug 2022

A novel lineage of the Capra genus discovered in the Taurus Mountains of Turkey using ancient genomicsGoat ancient DNA analysis unveils a new lineage that may have hybridized with domestic goatsRecommended by Laura Botigué based on reviews by Torsten Günther and 1 anonymous reviewerThe genomic analysis of ancient remains has revolutionized the study of the past over the last decade. On top of the discoveries related to human evolution, plant and animal archaeogenomics has been used to gain new insights into the domestication process and the dispersal of domestic forms. In this study, Daly and colleagues analyse the genomic data from seven goat specimens from the Epipalaeolithic recovered from the Direkli Cave in the Taurus Mountains in southern Turkey. They also generate new genomic data from Capra lineages across the phylogeny, contributing to the availability of genomic resources for this genus. Analysis of the ancient remains is compared to modern genomic variability and sheds light on the complexity of the Tur wild Capra lineages and their relationship with domestic goats and their wild ancestors. Authors find that during the Late Pleistocene in the Taurus Mountains wild goats from the Tur lineage, today restricted to the Caucasus region, were not rare and cohabited with Bezoar, the wild goats that are the ancestors of domestic goats. They identify the Direkli Cave specimens as a lineage separate from the A modified D statistic, Dex, is developed to examine the contribution of the ancient Tur lineage in domestic goats through time and space. Dex measures the relative degree of allele sharing, derived specifically in a selected genome or group of genomes, and may have some utility in genera with complex admixture histories or admixture from ghost lineages. Results confirm that Neolithic European goat had an excess of allele sharing with this ancient Tur lineage, something that is absent in contemporary goats eastwards or in modern goats. Interspecific gene flow is not uncommon among mammals, but the case of Capra has the additional motivation of understanding the origins of the domestic species. This work uncovers an ancient Tur lineage that is different from the modern ones and is additionally found in another geographic area. Furthermore, evidence shows that this ancient lineage exhibits substantial amounts of allele sharing with the wild ancestor of the domestic goat, but also with the Neolithic Eurasian domestic goats, highlighting the complexity of the domestication process. This work has also important implications in understanding the effect of over-hunting and habitat disruption during the Anthropocene on the evolution of the Capra genus. The availability of more ancient specimens and better coverage of the modern genomic variability can help quantifying the lineages that went lost and identify the causes of their extinction. This work is limited by the current availability of whole genomes from modern Capra specimens, but pieces of evidence as well that an effort is needed to obtain more genomic data from ancient goats from different geographic ranges to determine to what extent these lineages contributed to goat domestication. References Daly KG, Arbuckle BS, Rossi C, Mattiangeli V, Lawlor PA, Mashkour M, Sauer E, Lesur J, Atici L, Cevdet CM and Bradley DG (2022) A novel lineage of the Capra genus discovered in the Taurus Mountains of Turkey using ancient genomics. bioRxiv, 2022.04.08.487619, ver. 5 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2022.04.08.487619 | A novel lineage of the Capra genus discovered in the Taurus Mountains of Turkey using ancient genomics | Kevin G. Daly, Benjamin S. Arbuckle, Conor Rossi, Valeria Mattiangeli, Phoebe A. Lawlor, Marjan Mashkour, Eberhard Sauer, Joséphine Lesur, Levent Atici, Cevdet Merih Erek, Daniel G. Bradley | <p>Direkli Cave, located in the Taurus Mountains of southern Turkey, was occupied by Late Epipaleolithic hunters-gatherers for the seasonal hunting and processing of game including large numbers of wild goats. We report genomic data from new and p... | | Evolutionary genomics, Population genomics, Vertebrates | Laura Botigué | 2022-04-15 12:05:47 | ||
11 May 2024
The European Reference Genome Atlas: piloting a decentralised approach to equitable biodiversity genomicsInformed Choices, Cohesive Future: Decisions and Recommendations for ERGARecommended by Jitendra Narayan based on reviews by Justin Ideozu and Eric Crandall
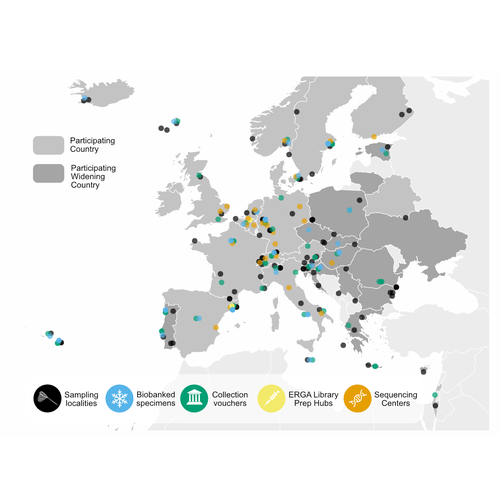
The European Reference Genome Atlas (ERGA) (Mc Cartney et al, 2024, Mazzoni et al, 2023) demonstrates the collaborative spirit and intellectual abilities of researchers from 33 European countries. This ambitious project, which is part of the Earth BioGenome Project (Lewin et al., 2018) Phase II, has embarked on an unprecedented mission: to decipher the genetic makeup of 150,000 species over a span of four years. At the heart of ERGA is a decentralized pilot infrastructure specifically built to assist the production of high-quality reference genomes. This structure acts as a scaffold for the massive task of genome sequencing, giving the necessary framework to manage the complexity of genomic research. The research paper under consideration offers a comprehensive narrative of ERGA's evolution, outlining both successes and challenges encountered along the road. One of the most significant issues addressed in the manuscript is the equitable distribution of resources and expertise among participating laboratories and countries. In a project of this magnitude, it is critical to leverage the pooled talents and capacities of researchers from across Europe. ERGA's pan-European network promotes communications and collaboration, creating an environment in which knowledge flows freely and barriers are overcome. This adoption of strong coordination and communication tactics will be essential to ERGA's success. Scientific collaboration depends on efficient communication channels because they allow researchers to share resources, collaborate on new initiatives, and exchange ideas. Through a diverse range of gatherings, courses, and virtual discussion boards, ERGA fosters an environment of transparency and cooperation among members, enabling scientists to overcome challenges and make significant discoveries. The importance ERGA places on training and information transfer programmes is a pillar of its strategy. Understanding the importance of capacity development, ERGA invests in providing researchers with the knowledge and abilities necessary for effectively navigating the complicated terrain of genomic research. A wide range of subjects are covered in training programmes (Larivière et al. 2023), from sample preparation and collection to data processing methods and sequencing technology. Through the development of a group of highly qualified experts, ERGA creates the foundation for continued advancement and creativity in the genomics sector. This manuscript also covers in detail the technological workflows and sequencing techniques used in ERGA's pilot infrastructure. With the aid of cutting-edge sequencing technologies based on both long-read and short-read sequencing, they are working to unravel the complex structure of the genetic code with a level of accuracy and precision never before possible. To guarantee the accuracy of genetic data and prevent mistakes and flaws that can jeopardize the findings' integrity, quality control methods are put in place. Despite having a focus on genome sequencing due to its technological complexities, ERGA also remains firm in its dedication to metadata collection and sample validation. Metadata serves as a critical link between raw genetic data and useful scientific insights, giving necessary context and allowing researchers to draw practical findings from their investigations. Sample validation approaches improve the reliability and reproducibility of the results, providing users confidence in the quality of the genetic data provided by ERGA. Looking ahead, ERGA envisions its decentralized infrastructure serving as a model for global collaborative research efforts. By embracing diversity, encouraging cooperation, and pushing for open access to data and resources, ERGA hopes to catalyze scientific discovery and generate positive change in the field of biodiversity genomics. ERGA aims to promote a more equitable and sustainable future for all by ongoing interaction with stakeholders, intensive outreach and education activities, and policy change advocacy. In addition to its immediate goals, ERGA considers the long-term implications of its work. As genomic technology progresses, the potential application of high-quality reference genomes will continue to grow. From informing conservation efforts and illuminating evolutionary histories to revolutionizing healthcare and agriculture, it is likely that ERGA's contributions will have far-reaching consequences for people and the planet as a whole. Furthermore, ERGA understands the importance of interdisciplinary collaboration in addressing the difficult challenges of the twenty-first century. ERGA aims to integrate genetic research into larger initiatives to promote sustainability and biodiversity conservation by forming relationships with stakeholders from other areas, such as policymakers, conservationists, and indigenous groups. Through shared knowledge and community action, ERGA seeks to create a future in which mankind coexists peacefully with the natural world, guided by a thorough grasp of its genetic legacy and ecological interconnectivity. Finally, the manuscript exemplifies ERGA's collaborative ambitions and achievements, capturing the spirit of creativity and collaboration that defines this ground-breaking effort. As ERGA continues to push the boundaries of genetic research, it remains dedicated to scientific excellence, inclusivity, and the quest of knowledge for the benefit of society. I wholeheartedly recommend the publication of this groundbreaking initiative, offering my enthusiastic endorsement for its valuable contribution to the scientific community. References Lewin, H. A., Robinson, G. E., Kress, W. J., Baker, W. J., Coddington, J., Crandall, K. A., Durbin, R., Edwards, S. V., Forest, F., Gilbert, M. T. P., Goldstein, M. M., Grigoriev, I. V., Hackett, K. J., Haussler, D., Jarvis, E. D., Johnson, W. E., Patrinos, A., Richards, S., Castilla-Rubio, J. C., … Zhang, G. (2018). Earth BioGenome Project: Sequencing life for the future of life. Proceedings of the National Academy of Sciences, 115(17), 4325–4333. https://doi.org/10.1073/pnas.1720115115 Mazzoni, C. J., Claudio, C.i, Waterhouse, R. M. (2023). Biodiversity: an atlas of European reference genomes. Nature 619 : 252-252. https://doi.org/10.1038/d41586-023-02229-w Mc Cartney, A. M., Formenti, G., Mouton, A., Panis, D. de, Marins, L. S., Leitão, H. G., Diedericks, G., Kirangwa, J., Morselli, M., Salces-Ortiz, J., Escudero, N., Iannucci, A., Natali, C., Svardal, H., Fernández, R., Pooter, T. de, Joris, G., Strazisar, M., Wood, J., … Mazzoni, C. J. (2024). The European Reference Genome Atlas: piloting a decentralised approach to equitable biodiversity genomics. bioRxiv, ver. 4 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2023.09.25.559365 | The European Reference Genome Atlas: piloting a decentralised approach to equitable biodiversity genomics | Ann M Mc Cartney, Giulio Formenti, Alice Mouton, Claudio Ciofi, Robert M Waterhouse, Camila J Mazzoni, Diego De Panis, Luisa S Schlude Marins, Henrique G Leitao, Genevieve Diedericks, Joseph Kirangwa, Marco Morselli, Judit Salces, Nuria Escudero, ... | <p>English: A global genome database of all of Earth's species diversity could be a treasure trove of scientific discoveries. However, regardless of the major advances in genome sequencing technologies, only a tiny fraction of species have genomic... | | Bioinformatics, ERGA Pilot | Jitendra Narayan | Justin Ideozu, Eric Crandall | 2023-10-01 01:03:58 | |
09 Oct 2020
An evaluation of pool-sequencing transcriptome-based exon capture for population genomics in non-model speciesAssessing a novel sequencing-based approach for population genomics in non-model speciesRecommended by Thomas Derrien and Sebastian Ernesto Ramos-Onsins based on reviews by Valentin Wucher and 1 anonymous reviewer
Developing new sequencing and bioinformatic strategies for non-model species is of great interest in many applications, such as phylogenetic studies of diverse related species, but also for studies in population genomics, where a relatively large number of individuals is necessary. Different approaches have been developed and used in these last two decades, such as RAD-Seq (e.g., Miller et al. 2007), exome sequencing (e.g., Teer and Mullikin 2010) and other genome reduced representation methods that avoid the use of a good reference and well annotated genome (reviewed at Davey et al. 2011). However, population genomics studies require the analysis of numerous individuals, which makes the studies still expensive. Pooling samples was thought as an inexpensive strategy to obtain estimates of variability and other related to the frequency spectrum, thus allowing the study of variability at population level (e.g., Van Tassell et al. 2008), although the major drawback was the loss of information related to the linkage of the variants. In addition, population analysis using all these sequencing strategies require statistical and empirical validations that are not always fully performed. A number of studies aiming to obtain unbiased estimates of variability using reduced representation libraries and/or with pooled data have been performed (e.g., Futschik and Schlötterer 2010, Gautier et al. 2013, Ferretti et al. 2013, Lynch et al. 2014), as well as validation of new sequencing methods for population genetic analyses (e.g., Gautier et al. 2013, Nevado et al. 2014). Nevertheless, empirical validation using both pooled and individual experimental approaches combined with different bioinformatic methods has not been always performed. References [1] Choquet et al. (2019). Towards population genomics in non-model species with large genomes: a case study of the marine zooplankton Calanus finmarchicus. Royal Society open science, 6(2), 180608. doi: https://doi.org/10.1098/rsos.180608 | An evaluation of pool-sequencing transcriptome-based exon capture for population genomics in non-model species | Emeline Deleury, Thomas Guillemaud, Aurélie Blin & Eric Lombaert | <p>Exon capture coupled to high-throughput sequencing constitutes a cost-effective technical solution for addressing specific questions in evolutionary biology by focusing on expressed regions of the genome preferentially targeted by selection. Tr... | | Bioinformatics, Population genomics | Thomas Derrien | 2020-02-26 09:21:11 |





