
DERRIEN Thomas
- IGDR - Institute for Genetics and Development, CNRS, Rennes, France
- Bioinformatics, Functional genomics, Structural genomics
- recommender
Recommendation: 1
Reviews: 0
Recommendation: 1
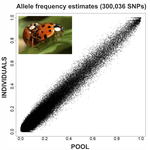
An evaluation of pool-sequencing transcriptome-based exon capture for population genomics in non-model species
Assessing a novel sequencing-based approach for population genomics in non-model species
Recommended by Thomas Derrien and Sebastian Ernesto Ramos-Onsins based on reviews by Valentin Wucher and 1 anonymous reviewerDeveloping new sequencing and bioinformatic strategies for non-model species is of great interest in many applications, such as phylogenetic studies of diverse related species, but also for studies in population genomics, where a relatively large number of individuals is necessary. Different approaches have been developed and used in these last two decades, such as RAD-Seq (e.g., Miller et al. 2007), exome sequencing (e.g., Teer and Mullikin 2010) and other genome reduced representation methods that avoid the use of a good reference and well annotated genome (reviewed at Davey et al. 2011). However, population genomics studies require the analysis of numerous individuals, which makes the studies still expensive. Pooling samples was thought as an inexpensive strategy to obtain estimates of variability and other related to the frequency spectrum, thus allowing the study of variability at population level (e.g., Van Tassell et al. 2008), although the major drawback was the loss of information related to the linkage of the variants. In addition, population analysis using all these sequencing strategies require statistical and empirical validations that are not always fully performed. A number of studies aiming to obtain unbiased estimates of variability using reduced representation libraries and/or with pooled data have been performed (e.g., Futschik and Schlötterer 2010, Gautier et al. 2013, Ferretti et al. 2013, Lynch et al. 2014), as well as validation of new sequencing methods for population genetic analyses (e.g., Gautier et al. 2013, Nevado et al. 2014). Nevertheless, empirical validation using both pooled and individual experimental approaches combined with different bioinformatic methods has not been always performed.
Here, Deleury et al. (2020) proposed an efficient and elegant way of quantifying the single-nucleotide polymorphisms (SNPs) of exon-derived sequences in a non-model species (i.e. for which no reference genome sequence is available) at the population level scale. They also designed a new procedure to capture exon-derived sequences based on a reference transcriptome. In addition, they were able to make predictions of intron-exon boundaries for de novo transcripts based on the decay of read depth at the ends of the coding regions.
Based on theoretical predictions (Gautier et al. 2013), Deleury et al. (2020) designed a procedure to test the accuracy of variant allele frequencies (AFs) with pooled samples, in a reduced genome-sequence library made with transcriptome regions, and additionally testing the effects of new bioinformatic methods in contrast to standardized methods. They applied their strategy on the non-model species Asian ladybird (Harmonia axyridis), for which a draft genome is available, thereby allowing them to benchmark their method with regard to a traditional mapping-based approach. Based on species-specific de novo transcriptomes, they designed capture probes which are then used to call SNPx and then compared the resulting SNP AFs at the individual (multiplexed) versus population (pooled) levels. Interestingly, they showed that SNP AFs in the pool sequencing strategy nicely correlate with the individual ones but obviously in a cost-effective way. Studies of population genomics for non-model species have usually limited budgets. The number of individuals required for population genomics analysis multiply the costs of the project, making pooling samples an interesting option. Furthermore, the use of pool sequencing is not always a choice, as many organisms are too small and/or individuals are too sticked each other to be individually sequenced (e.g., Choquet et al. 2019, Kurland et al. 2019). In addition, the study of a reduced section of the genome is cheaper and often sufficient for a number of population genetic questions, such as the understanding of general demographic events, or the estimation of the effects of positive and/or negative selection at functional coding regions. Studies on population genomics of non-model species have many applications in related fields, such as conservation genetics, control of invasive species, etc. The work of Deleury et al. (2020) is an elegant contribution to the assessment and validation of new methodologies used for the analysis of genome variations at the intra-population variability level, highlighting straight bioinformatic and reliable sequencing methods for population genomics studies.
References
[1] Choquet et al. (2019). Towards population genomics in non-model species with large genomes: a case study of the marine zooplankton Calanus finmarchicus. Royal Society open science, 6(2), 180608. doi: https://doi.org/10.1098/rsos.180608
[2] Davey, J. W., Hohenlohe, P. A., Etter, P. D., Boone, J. Q., Catchen, J. M. and Blaxter, M. L. (2011). Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nature Reviews Genetics, 12(7), 499-510. doi: https://doi.org/10.1038/nrg3012
[3] Deleury, E., Guillemaud, T., Blin, A. and Lombaert, E. (2020) An evaluation of pool-sequencing transcriptome-based exon capture for population genomics in non-model species. bioRxiv, 10.1101/583534, ver. 7 peer-reviewed and recommended by PCI Genomics. https://doi.org/10.1101/583534
[4] Ferretti, L., Ramos‐Onsins, S. E. and Pérez‐Enciso, M. (2013). Population genomics from pool sequencing. Molecular ecology, 22(22), 5561-5576. doi: https://doi.org/10.1111/mec.12522
[5] Futschik, A. and Schlötterer, C. (2010). Massively parallel sequencing of pooled DNA samples—the next generation of molecular markers. Genetics, 186 (1), 207-218. doi: https://doi.org/10.1534/genetics.110.114397
[6] Gautier et al. (2013). Estimation of population allele frequencies from next‐generation sequencing data: pool‐versus individual‐based genotyping. Molecular Ecology, 22(14), 3766-3779. doi: https://doi.org/10.1111/mec.12360
[7] Kurland et al. (2019). Exploring a Pool‐seq‐only approach for gaining population genomic insights in nonmodel species. Ecology and evolution, 9(19), 11448-11463. doi: https://doi.org/10.1002/ece3.5646
[8] Lynch, M., Bost, D., Wilson, S., Maruki, T. and Harrison, S. (2014). Population-genetic inference from pooled-sequencing data. Genome biology and evolution, 6(5), 1210-1218. doi: https://doi.org/10.1093/gbe/evu085
[9] Miller, M. R., Dunham, J. P., Amores, A., Cresko, W. A. and Johnson, E. A. (2007). Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers. Genome research, 17(2), 240-248. doi: https://doi.org/10.1101%2Fgr.5681207
[10] Nevado, B., Ramos‐Onsins, S. E. and Perez‐Enciso, M. (2014). Resequencing studies of nonmodel organisms using closely related reference genomes: optimal experimental designs and bioinformatics approaches for population genomics. Molecular ecology, 23(7), 1764-1779. doi: https://doi.org/10.1111/mec.12693
[11] Teer, J. K. and Mullikin, J. C. (2010). Exome sequencing: the sweet spot before whole genomes. Human molecular genetics, 19(R2), R145-R151. doi: https://doi.org/10.1093/hmg/ddq333
[12] Van Tassell et al. (2008). SNP discovery and allele frequency estimation by deep sequencing of reduced representation libraries. Nature methods, 5(3), 247-252. doi: https://doi.org/10.1038/nmeth.1185