
Latest recommendations

| Id | Title | Authors | Abstract | Picture▲ | Thematic fields | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
02 Jun 2023
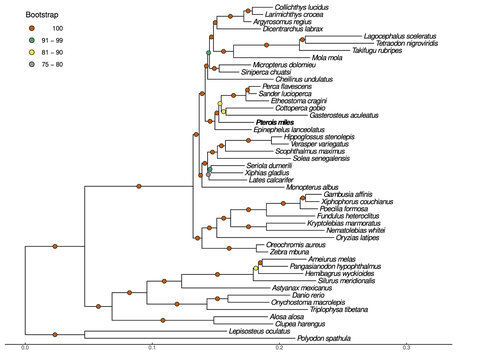
Near-chromosome level genome assembly of devil firefish, Pterois milesThe genome of a dangerous invader (fish) beautyRecommended by Iker Irisarri based on reviews by Maria Recuerda and 1 anonymous reviewer based on reviews by Maria Recuerda and 1 anonymous reviewer
High-quality genomes are currently being generated at an unprecedented speed powered by long-read sequencing technologies. However, sequencing effort is concentrated unequally across the tree of life and several key evolutionary and ecological groups remain largely unexplored. So is the case for fish species of the family Scorpaenidae (Perciformes). Kitsoulis et al. present the genome of the devil firefish, Pterois miles (1). Following current best practices, the assembly relies largely on Oxford Nanopore long reads, aided by Illumina short reads for polishing to increase the per-base accuracy. PacBio’s IsoSeq was used to sequence RNA from a variety of tissues as direct evidence for annotating genes. The reconstructed genome is 902 Mb in size and has high contiguity (N50=14.5 Mb; 660 scaffolds, 90% of the genome covered by the 83 longest scaffolds) and completeness (98% BUSCO completeness). The new genome is used to assess the phylogenetic position of P. miles, explore gene synteny against zebrafish, look at orthogroup expansion and contraction patterns in Perciformes, as well as to investigate the evolution of toxins in scorpaenid fish (2). In addition to its value for better understanding the evolution of scorpaenid and teleost fishes, this new genome is also an important resource for monitoring its invasiveness through the Mediterranean Sea (3) and the Atlantic Ocean, in the latter case forming the invasive lionfish complex with P. volitans (4). REFERENCES 1. Kitsoulis CV, Papadogiannis V, Kristoffersen JB, Kaitetzidou E, Sterioti E, Tsigenopoulos CS, Manousaki T. (2023) Near-chromosome level genome assembly of devil firefish, Pterois miles. BioRxiv, ver. 6 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2023.01.10.523469 2. Kiriake A, Shiomi K. (2011) Some properties and cDNA cloning of proteinaceous toxins from two species of lionfish (Pterois antennata and Pterois volitans). Toxicon, 58(6-7):494–501. https://doi.org/10.1016/j.toxicon.2011.08.010 3. Katsanevakis S, et al. (2020) Un- published Mediterranean records of marine alien and cryptogenic species. BioInvasions Records, 9:165–182. https://doi.org/10.3391/bir.2020.9.2.01 4. Lyons TJ, Tuckett QM, Hill JE. (2019) Data quality and quantity for invasive species: A case study of the lionfishes. Fish and Fisheries, 20:748–759. https://doi.org/10.1111/faf.12374 | Near-chromosome level genome assembly of devil firefish, *Pterois miles* | Christos V. Kitsoulis, Vasileios Papadogiannis, Jon B. Kristoffersen, Elisavet Kaitetzidou, Aspasia Sterioti, Costas S. Tsigenopoulos, Tereza Manousaki | <p style="text-align: justify;">Devil firefish (<em>Pterois miles</em>), a member of Scorpaenidae family, is one of the most successful marine non-native species, dominating around the world, that was rapidly spread into the Mediterranean Sea, thr... | | Evolutionary genomics | Iker Irisarri | 2023-01-17 12:37:20 | ||
02 Apr 2021

Semi-artificial datasets as a resource for validation of bioinformatics pipelines for plant virus detectionToward a critical assessment of virus detection in plantsRecommended by Hadi Quesneville based on reviews by Alexander Suh and 1 anonymous reviewerThe advent of High Throughput Sequencing (HTS) since the last decade has revealed previously unsuspected diversity of viruses as well as their (sometimes) unexpected presence in some healthy individuals. These results demonstrate that genomics offers a powerful tool for studying viruses at the individual level, allowing an in-depth inventory of those that are infecting an organism. Such approaches make it possible to study viromes with an unprecedented level of detail, both qualitative and quantitative, which opens new venues for analyses of viruses of humans, animals and plants. Consequently, the diagnostic field is using more and more HTS, fueling the need for efficient and reliable bioinformatics tools. Many such tools have already been developed, but in plant disease diagnostics, validation of the bioinformatics pipelines used for the detection of viruses in HTS datasets is still in its infancy. There is an urgent need for benchmarking the different tools and algorithms using well-designed reference datasets generated for this purpose. This is a crucial step to move forward and to improve existing solutions toward well-standardized bioinformatics protocols. This context has led to the creation of the Plant Health Bioinformatics Network (PHBN), a Euphresco network project aiming to build a bioinformatics community working on plant health. One of their objectives is to provide researchers with open-access reference datasets allowing to compare and validate virus detection pipelines. In this framework, Tamisier et al. [1] present real, semi-artificial, and completely artificial datasets, each aimed at addressing challenges that could affect virus detection. These datasets comprise real RNA-seq reads from virus-infected plants as well as simulated virus reads. Such a work, providing open-access datasets for benchmarking bioinformatics tools, should be encouraged as they are key to software improvement as demonstrated by the well-known success story of the protein structure prediction community: their pioneer community-wide effort, called Critical Assessment of protein Structure Prediction (CASP)[2], has been providing research groups since 1994 with an invaluable way to objectively test their structure prediction methods, thereby delivering an independent assessment of state-of-art protein-structure modelling tools. Following this success, many other bioinformatic community developed similar “competitions”, such as RNA-puzzles [3] to predict RNA structures, Critical Assessment of Function Annotation [4] to predict gene functions, Critical Assessment of Prediction of Interactions [5] to predict protein-protein interactions, Assemblathon [6] for genome assembly, etc. These are just a few examples from a long list of successful initiatives. Such efforts enable rigorous assessments of tools, stimulate the developers’ creativity, but also provide user communities with a state-of-art evaluation of available tools. Inspired by these success stories, the authors propose a “VIROMOCK challenge” [7], asking researchers in the field to test their tools and to provide feedback on each dataset through a repository. This initiative, if well followed, will undoubtedly improve the field of virus detection in plants, but also probably in many other organisms. This will be a major contribution to the field of viruses, leading to better diagnostics and, consequently, a better understanding of viral diseases, thus participating in promoting human, animal and plant health. References [1] Tamisier, L., Haegeman, A., Foucart, Y., Fouillien, N., Al Rwahnih, M., Buzkan, N., Candresse, T., Chiumenti, M., De Jonghe, K., Lefebvre, M., Margaria, P., Reynard, J.-S., Stevens, K., Kutnjak, D. and Massart, S. (2021) Semi-artificial datasets as a resource for validation of bioinformatics pipelines for plant virus detection. Zenodo, 4273791, version 4 peer-reviewed and recommended by Peer community in Genomics. doi: https://doi.org/10.5281/zenodo.4273791 [2] Critical Assessment of protein Structure Prediction” (CASP) - https://en.wikipedia.org/wiki/CASP [3] RNA-puzzles - https://www.rnapuzzles.org [4] Critical Assessment of Function Annotation (CAFA) - https://en.wikipedia.org/wiki/Critical_Assessment_of_Function_Annotation [5] Critical Assessment of Prediction of Interactions (CAPI) - https://en.wikipedia.org/wiki/Critical_Assessment_of_Prediction_of_Interactions [6] Assemblathon - https://assemblathon.org [7] VIROMOCK challenge - https://gitlab.com/ilvo/VIROMOCKchallenge | Semi-artificial datasets as a resource for validation of bioinformatics pipelines for plant virus detection | Lucie Tamisier, Annelies Haegeman, Yoika Foucart, Nicolas Fouillien, Maher Al Rwahnih, Nihal Buzkan, Thierry Candresse, Michela Chiumenti, Kris De Jonghe, Marie Lefebvre, Paolo Margaria, Jean Sébastien Reynard, Kristian Stevens, Denis Kutnjak, Séb... | <p>The widespread use of High-Throughput Sequencing (HTS) for detection of plant viruses and sequencing of plant virus genomes has led to the generation of large amounts of data and of bioinformatics challenges to process them. Many bioinformatics... | | Bioinformatics, Plants, Viruses and transposable elements | Hadi Quesneville | 2020-11-27 14:31:47 | ||
05 May 2021
A primer and discussion on DNA-based microbiome data and related bioinformatics analysesA hitchhiker’s guide to DNA-based microbiome analysisRecommended by Danny Ionescu based on reviews by Nicolas Pollet, Rafael Cuadrat and 1 anonymous reviewer
In the last two decades, microbial research in its different fields has been increasingly focusing on microbiome studies. These are defined as studies of complete assemblages of microorganisms in given environments and have been benefiting from increases in sequencing length, quality, and yield, coupled with ever-dropping prices per sequenced nucleotide. Alongside localized microbiome studies, several global collaborative efforts have emerged, including the Human Microbiome Project [1], the Earth Microbiome Project [2], the Extreme Microbiome Project, and MetaSUB [3]. Coupled with the development of sequencing technologies and the ever-increasing amount of data output, multiple standalone or online bioinformatic tools have been designed to analyze these data. Often these tools have been focusing on either of two main tasks: 1) Community analysis, providing information on the organisms present in the microbiome, or 2) Functionality, in the case of shotgun metagenomic data, providing information on the metabolic potential of the microbiome. Bridging between the two types of data, often extracted from the same dataset, is typically a daunting task that has been addressed by a handful of tools only. The extent of tools and approaches to analyze microbiome data is great and may be overwhelming to researchers new to microbiome or bioinformatic studies. In their paper “A primer and discussion on DNA-based microbiome data and related bioinformatics analyses”, Douglas and Langille [4] guide us through the different sequencing approaches useful for microbiome studies. alongside their advantages and caveats and a selection of tools to analyze these data, coupled with examples from their own field of research. Standing out in their primer-style review is the emphasis on the coupling between taxonomic/phylogenetic identification of the organisms and their functionality. This type of analysis, though highly important to understand the role of different microorganisms in an environment as well as to identify potential functional redundancy, is often not conducted. For this, the authors identify two approaches. The first, using shotgun metagenomics, has higher chances of attributing a function to the correct taxon. The second, using amplicon sequencing of marker genes, allows for a deeper coverage of the microbiome at a lower cost, and extrapolates the amplicon data to close relatives with a sequenced genome. As clearly stated, this approach makes the leap between taxonomy and functionality and has been shown to be erroneous in cases where the core genome of the bacterial genus or family does not encompass the functional diversity of the different included species. This practice was already common before the genomic era, but its accuracy is improving thanks to the increasing availability of sequenced reference genomes from cultures, environmentally picked single cells or metagenome-assembled genome. In addition to their description of standalone tools useful for linking taxonomy and functionality, one should mention the existence of online tools that may appeal to researchers who do not have access to adequate bioinformatics infrastructure. Among these are the Integrated Microbial Genomes and Microbiomes (IMG) from the Joint Genome Institute [5], KBase [6] and MG-RAST [7]. A second important point arising from this review is the need for standardization in microbiome data analyses and the complexity of achieving this. As Douglas and Langille [4] state, this has been previously addressed, highlighting the variability in results obtained with different tools. It is often the case that papers describing new bioinformatic tools display their superiority relative to existing alternatives, potentially misleading newcomers to the field that the newest tool is the best and only one to be used. This is often not the case, and while benchmarking against well-defined datasets serves as a powerful testing tool, “real-life” samples are often not comparable. Thus, as done here, future primer-like reviews should highlight possible cross-field caveats, encouraging researchers to employ and test several approaches and validate their results whenever possible. In summary, Douglas and Langille [4] offer both the novice and experienced researcher a detailed guide along the paths of microbiome data analysis, accompanied by informative background information, suggested tools with which analyses can be started, and an insightful view on where the field should be heading. References [1] Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI (2007) The Human Microbiome Project. Nature, 449, 804–810. https://doi.org/10.1038/nature06244 [2] Gilbert JA, Jansson JK, Knight R (2014) The Earth Microbiome project: successes and aspirations. BMC Biology, 12, 69. https://doi.org/10.1186/s12915-014-0069-1 [3] Mason C, Afshinnekoo E, Ahsannudin S, Ghedin E, Read T, Fraser C, Dudley J, Hernandez M, Bowler C, Stolovitzky G, Chernonetz A, Gray A, Darling A, Burke C, Łabaj PP, Graf A, Noushmehr H, Moraes s., Dias-Neto E, Ugalde J, Guo Y, Zhou Y, Xie Z, Zheng D, Zhou H, Shi L, Zhu S, Tang A, Ivanković T, Siam R, Rascovan N, Richard H, Lafontaine I, Baron C, Nedunuri N, Prithiviraj B, Hyat S, Mehr S, Banihashemi K, Segata N, Suzuki H, Alpuche Aranda CM, Martinez J, Christopher Dada A, Osuolale O, Oguntoyinbo F, Dybwad M, Oliveira M, Fernandes A, Oliveira M, Fernandes A, Chatziefthimiou AD, Chaker S, Alexeev D, Chuvelev D, Kurilshikov A, Schuster S, Siwo GH, Jang S, Seo SC, Hwang SH, Ossowski S, Bezdan D, Udekwu K, Udekwu K, Lungjdahl PO, Nikolayeva O, Sezerman U, Kelly F, Metrustry S, Elhaik E, Gonnet G, Schriml L, Mongodin E, Huttenhower C, Gilbert J, Hernandez M, Vayndorf E, Blaser M, Schadt E, Eisen J, Beitel C, Hirschberg D, Schriml L, Mongodin E, The MetaSUB International Consortium (2016) The Metagenomics and Metadesign of the Subways and Urban Biomes (MetaSUB) International Consortium inaugural meeting report. Microbiome, 4, 24. https://doi.org/10.1186/s40168-016-0168-z [4] Douglas GM, Langille MGI (2021) A primer and discussion on DNA-based microbiome data and related bioinformatics analyses. OSF Preprints, ver. 4 peer-reviewed and recommended by Peer Community In Genomics. https://doi.org/10.31219/osf.io/3dybg [5] Chen I-MA, Markowitz VM, Chu K, Palaniappan K, Szeto E, Pillay M, Ratner A, Huang J, Andersen E, Huntemann M, Varghese N, Hadjithomas M, Tennessen K, Nielsen T, Ivanova NN, Kyrpides NC (2017) IMG/M: integrated genome and metagenome comparative data analysis system. Nucleic Acids Research, 45, D507–D516. https://doi.org/10.1093/nar/gkw929 [6] Arkin AP, Cottingham RW, Henry CS, Harris NL, Stevens RL, Maslov S, Dehal P, Ware D, Perez F, Canon S, Sneddon MW, Henderson ML, Riehl WJ, Murphy-Olson D, Chan SY, Kamimura RT, Kumari S, Drake MM, Brettin TS, Glass EM, Chivian D, Gunter D, Weston DJ, Allen BH, Baumohl J, Best AA, Bowen B, Brenner SE, Bun CC, Chandonia J-M, Chia J-M, Colasanti R, Conrad N, Davis JJ, Davison BH, DeJongh M, Devoid S, Dietrich E, Dubchak I, Edirisinghe JN, Fang G, Faria JP, Frybarger PM, Gerlach W, Gerstein M, Greiner A, Gurtowski J, Haun HL, He F, Jain R, Joachimiak MP, Keegan KP, Kondo S, Kumar V, Land ML, Meyer F, Mills M, Novichkov PS, Oh T, Olsen GJ, Olson R, Parrello B, Pasternak S, Pearson E, Poon SS, Price GA, Ramakrishnan S, Ranjan P, Ronald PC, Schatz MC, Seaver SMD, Shukla M, Sutormin RA, Syed MH, Thomason J, Tintle NL, Wang D, Xia F, Yoo H, Yoo S, Yu D (2018) KBase: The United States Department of Energy Systems Biology Knowledgebase. Nature Biotechnology, 36, 566–569. https://doi.org/10.1038/nbt.4163 [7] Wilke A, Bischof J, Gerlach W, Glass E, Harrison T, Keegan KP, Paczian T, Trimble WL, Bagchi S, Grama A, Chaterji S, Meyer F (2016) The MG-RAST metagenomics database and portal in 2015. Nucleic Acids Research, 44, D590–D594. https://doi.org/10.1093/nar/gkv1322 | A primer and discussion on DNA-based microbiome data and related bioinformatics analyses | Gavin M. Douglas and Morgan G. I. Langille | <p style="text-align: justify;">The past decade has seen an eruption of interest in profiling microbiomes through DNA sequencing. The resulting investigations have revealed myriad insights and attracted an influx of researchers to the research are... | | Bioinformatics, Metagenomics | Danny Ionescu | 2021-02-17 00:26:46 | ||
25 Nov 2022
Phenotypic and transcriptomic analyses reveal major differences between apple and pear scab nonhost resistanceApples and pears: two closely related species with differences in scab nonhost resistanceRecommended by Wirulda Pootakham based on reviews by 3 anonymous reviewersNonhost resistance is a common form of disease resistance exhibited by plants against microorganisms that are pathogenic to other plant species [1]. Apples and pears are two closely related species belonging to Rosaceae family, both affected by scab disease caused by fungal pathogens in the Venturia genus. These pathogens appear to be highly host-specific. While apples are nonhosts for Venturia pyrina, pears are nonhosts for Venturia inaequalis. To date, the molecular bases of scab nonhost resistance in apple and pear have not been elucidated. This preprint by Vergne, et al (2022) [2] analyzed nonhost resistance symptoms in apple/V. pyrina and pear/V. inaequalis interactions as well as their transcriptomic responses. Interestingly, the author demonstrated that the nonhost apple/V. pyrina interaction was almost symptomless while hypersensitive reactions were observed for pear/V. inaequalis interaction. The transcriptomic analyses also revealed a number of differentially expressed genes (DEGs) that corresponded to the severity of the interactions, with very few DEGs observed during the apple/V. pyrina interaction and a much higher number of DEGs during the pear/V. inaequalis interaction. This type of reciprocal host-pathogen interaction study is valuable in gaining new insights into how plants interact with microorganisms that are potential pathogens in related species. A few processes appeared to be involved in the pear resistance against the nonhost pathogen V. inaequalis at the transcriptomic level, such as stomata closure, modification of cell wall and production of secondary metabolites as well as phenylpropanoids. Based on the transcriptomics changes during the nonhost interaction, the author compared the responses to those of host-pathogen interactions and revealed some interesting findings. They proposed a series of cascading effects in pear induced by the presence of V. inaequalis, which I believe helps shed some light on the basic mechanism for nonhost resistance. I am recommending this study because it provides valuable information that will strengthen our understanding of nonhost resistance in the Rosaceae family and other plant species. The knowledge gained here may be applied to genetically engineer plants for a broader resistance against a number of pathogens in the future. References 1. Senthil-Kumar M, Mysore KS (2013) Nonhost Resistance Against Bacterial Pathogens: Retrospectives and Prospects. Annual Review of Phytopathology, 51, 407–427. https://doi.org/10.1146/annurev-phyto-082712-102319 2. Vergne E, Chevreau E, Ravon E, Gaillard S, Pelletier S, Bahut M, Perchepied L (2022) Phenotypic and transcriptomic analyses reveal major differences between apple and pear scab nonhost resistance. bioRxiv, 2021.06.01.446506, ver. 4 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2021.06.01.446506 | Phenotypic and transcriptomic analyses reveal major differences between apple and pear scab nonhost resistance | E. Vergne, E. Chevreau, E. Ravon, S. Gaillard, S. Pelletier, M. Bahut, L. Perchepied | <p style="text-align: justify;"><strong>Background. </strong>Nonhost resistance is the outcome of most plant/pathogen interactions, but it has rarely been described in Rosaceous fruit species. Apple (<em>Malus x domestica</em> Borkh.) have a nonho... | | Functional genomics, Plants | Wirulda Pootakham | Jessica Soyer, Anonymous | 2022-05-13 15:06:08 | |
24 Jan 2024

High quality genome assembly of the brown hare (Lepus europaeus) with chromosome-level scaffoldingA high quality reference genome of the brown hareRecommended by Ed Hollox based on reviews by Merce Montoliu-Nerin and 1 anonymous reviewer
The brown hare, or European hare, Lupus europaeus, is a widespread mammal whose natural range spans western Eurasia. At the northern limit of its range, it hybridises with the mountain hare (L. timidis), and humans have introduced it into other continents. It represents a particularly interesting mammal to study for its population genetics, extensive hybridisation zones, and as an invasive species. This study (Michell et al. 2024) has generated a high-quality assembly of a genome from a brown hare from Finland using long PacBio HiFi sequencing reads and Hi-C scaffolding. The contig N50 of this new genome is 43 Mb, and completeness, assessed using BUSCO, is 96.1%. The assembly comprises 23 autosomes, and an X chromosome and Y chromosome, with many chromosomes including telomeric repeats, indicating the high level of completeness of this assembly. While the genome of the mountain hare has previously been assembled, its assembly was based on a short-read shotgun assembly, with the rabbit as a reference genome. The new high-quality brown hare genome assembly allows a direct comparison with the rabbit genome assembly. For example, the assembly addresses the karyotype difference between the hare (n=24) and the rabbit (n=22). Chromosomes 12 and 17 of the hare are equivalent to chromosome 1 of the rabbit, and chromosomes 13 and 16 of the hare are equivalent to chromosome 2 of the rabbit. The new assembly also provides a hare Y-chromosome, as the previous mountain hare genome was from a female. This new genome assembly provides an important foundation for population genetics and evolutionary studies of lagomorphs. References Michell, C., Collins, J., Laine, P. K., Fekete, Z., Tapanainen, R., Wood, J. M. D., Goffart, S., Pohjoismäki, J. L. O. (2024). High quality genome assembly of the brown hare (Lepus europaeus) with chromosome-level scaffolding. bioRxiv, ver. 3 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2023.08.29.555262 | High quality genome assembly of the brown hare (*Lepus europaeus*) with chromosome-level scaffolding | Craig Michell, Joanna Collins, Pia K. Laine, Zsofia Fekete, Riikka Tapanainen, Jonathan M. D. Wood, Steffi Goffart, Jaakko L. O. Pohjoismaki | <p style="text-align: justify;">We present here a high-quality genome assembly of the brown hare (Lepus europaeus Pallas), based on a fibroblast cell line of a male specimen from Liperi, Eastern Finland. This brown hare genome represents the first... | | ERGA Pilot, Vertebrates | Ed Hollox | 2023-10-16 20:46:39 | ||
09 Oct 2020
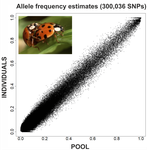
An evaluation of pool-sequencing transcriptome-based exon capture for population genomics in non-model speciesAssessing a novel sequencing-based approach for population genomics in non-model speciesRecommended by Thomas Derrien and Sebastian E. Ramos-Onsins based on reviews by Valentin Wucher and 1 anonymous reviewerDeveloping new sequencing and bioinformatic strategies for non-model species is of great interest in many applications, such as phylogenetic studies of diverse related species, but also for studies in population genomics, where a relatively large number of individuals is necessary. Different approaches have been developed and used in these last two decades, such as RAD-Seq (e.g., Miller et al. 2007), exome sequencing (e.g., Teer and Mullikin 2010) and other genome reduced representation methods that avoid the use of a good reference and well annotated genome (reviewed at Davey et al. 2011). However, population genomics studies require the analysis of numerous individuals, which makes the studies still expensive. Pooling samples was thought as an inexpensive strategy to obtain estimates of variability and other related to the frequency spectrum, thus allowing the study of variability at population level (e.g., Van Tassell et al. 2008), although the major drawback was the loss of information related to the linkage of the variants. In addition, population analysis using all these sequencing strategies require statistical and empirical validations that are not always fully performed. A number of studies aiming to obtain unbiased estimates of variability using reduced representation libraries and/or with pooled data have been performed (e.g., Futschik and Schlötterer 2010, Gautier et al. 2013, Ferretti et al. 2013, Lynch et al. 2014), as well as validation of new sequencing methods for population genetic analyses (e.g., Gautier et al. 2013, Nevado et al. 2014). Nevertheless, empirical validation using both pooled and individual experimental approaches combined with different bioinformatic methods has not been always performed. References [1] Choquet et al. (2019). Towards population genomics in non-model species with large genomes: a case study of the marine zooplankton Calanus finmarchicus. Royal Society open science, 6(2), 180608. doi: https://doi.org/10.1098/rsos.180608 | An evaluation of pool-sequencing transcriptome-based exon capture for population genomics in non-model species | Emeline Deleury, Thomas Guillemaud, Aurélie Blin & Eric Lombaert | <p>Exon capture coupled to high-throughput sequencing constitutes a cost-effective technical solution for addressing specific questions in evolutionary biology by focusing on expressed regions of the genome preferentially targeted by selection. Tr... | | Bioinformatics, Population genomics | Thomas Derrien | 2020-02-26 09:21:11 | ||
18 Jul 2022
CulebrONT: a streamlined long reads multi-assembler pipeline for prokaryotic and eukaryotic genomesA flexible and reproducible pipeline for long-read assembly and evaluationRecommended by Raúl Castanera based on reviews by Benjamin Istace and Valentine MurigneuxThird-generation sequencing has revolutionised de novo genome assembly. Thanks to this technology, genome reference sequences have evolved from fragmented drafts to gapless, telomere-to-telomere genome assemblies. Long reads produced by Oxford Nanopore and PacBio technologies can span structural variants and resolve complex repetitive regions such as centromeres, unlocking previously inaccessible genomic information. Nowadays, many research groups can afford to sequence the genome of their working model using long reads. Nevertheless, genome assembly poses a significant computational challenge. Read length, quality, coverage and genomic features such as repeat content can affect assembly contiguity, accuracy, and completeness in almost unpredictable ways. Consequently, there is no best universal software or protocol for this task. Producing a high-quality assembly requires chaining several tools into pipelines and performing extensive comparisons between the assemblies obtained by different tool combinations to decide which one is the best. This task can be extremely challenging, as the number of tools available rises very rapidly, and thorough benchmarks cannot be updated and published at such a fast pace. In their paper, Orjuela and collaborators present CulebrONT [1], a universal pipeline that greatly contributes to overcoming these challenges and facilitates long-read genome assembly for all taxonomic groups. CulebrONT incorporates six commonly used assemblers and allows to perform assembly, circularization (if needed), polishing, and evaluation in a simple framework. One important aspect of CulebrONT is its modularity, which allows the activation or deactivation of specific tools, giving great flexibility to the user. Nevertheless, possibly the best feature of CulebrONT is the opportunity to benchmark the selected tool combinations based on the excellent report generated by the pipeline. This HTML report aggregates the output of several tools for quality evaluation of the assemblies (e.g. BUSCO [2] or QUAST [3]) generated by the different assemblers, in addition to the running time and configuration parameters. Such information is of great help to identify the best-suited pipeline, as exemplified by the authors using four datasets of different taxonomic origins. Finally, CulebrONT can handle multiple samples in parallel, which makes it a good solution for laboratories looking for multiple assemblies on a large scale. References 1. Orjuela J, Comte A, Ravel S, Charriat F, Vi T, Sabot F, Cunnac S (2022) CulebrONT: a streamlined long reads multi-assembler pipeline for prokaryotic and eukaryotic genomes. bioRxiv, 2021.07.19.452922, ver. 5 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2021.07.19.452922 2. Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM (2015) BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics, 31, 3210–3212. https://doi.org/10.1093/bioinformatics/btv351 3. Gurevich A, Saveliev V, Vyahhi N, Tesler G (2013) QUAST: quality assessment tool for genome assemblies. Bioinformatics, 29, 1072–1075. https://doi.org/10.1093/bioinformatics/btt086 | CulebrONT: a streamlined long reads multi-assembler pipeline for prokaryotic and eukaryotic genomes | Julie Orjuela, Aurore Comte, Sébastien Ravel, Florian Charriat, Tram Vi, Francois Sabot, Sébastien Cunnac | <p style="text-align: justify;">Using long reads provides higher contiguity and better genome assemblies. However, producing such high quality sequences from raw reads requires to chain a growing set of tools, and determining the best workflow is ... | | Bioinformatics | Raúl Castanera | Valentine Murigneux | 2022-02-22 16:21:25 | |
07 Oct 2021

Fine-scale quantification of GC-biased gene conversion intensity in mammalsA systematic approach to the study of GC-biased gene conversion in mammalsRecommended by Carina Farah Mugal based on reviews by Fanny Pouyet , David Castellano and 1 anonymous reviewerThe role of GC-biased gene conversion (gBGC) in molecular evolution has interested scientists for the last two decades since its discovery in 1999 (Eyre-Walker 1999; Galtier et al. 2001). gBGC is a process that is associated with meiotic recombination, and is characterized by a transmission distortion in favor of G and C over A and T alleles at GC/AT heterozygous sites that occur in the vicinity of recombination-inducing double-strand breaks (Duret and Galtier 2009; Mugal et al. 2015). This transmission distortion results in a fixation bias of G and C alleles, equivalent to directional selection for G and C (Nagylaki 1983). The fixation bias subsequently leads to a correlation between recombination rate and GC content across the genome, which has served as indirect evidence for the prevalence of gBGC in many organisms. The fixation bias also produces shifts in the allele frequency spectrum (AFS) towards higher frequencies of G and C alleles. These molecular signatures of gBGC provide a means to quantify the strength of gBGC and study its variation among species and across the genome. Following this idea, first Lartillot (2013) and Capra et al. (2013) developed phylogenetic methodology to quantify gBGC based on substitutions, and De Maio et al. (2013) combined information on polymorphism into a phylogenetic setting. Complementary to the phylogenetic methods, later Glemin et al. (2015) developed a method that draws information solely from polymorphism data and the shape of the AFS. Application of these methods to primates (Capra et al. 2013; De Maio et al. 2013; Glemin et al. 2015) and mammals (Lartillot 2013) supported the notion that variation in the strength of gBGC across the genome reflects the dynamics of the recombination landscape, while variation among species correlates with proxies of the effective population size. However, application of the polymorphism-based method by Glemin et al. (2015) to distantly related Metazoa did not confirm the correlation with effective population size (Galtier et al. 2018). Here, Galtier (2021) introduces a novel phylogenetic approach applicable to the study of closely related species. Specifically, Galtier introduces a statistical framework that enables the systematic study of variation in the strength of gBGC among species and among genes. In addition, Galtier assesses fine-scale variation of gBGC across the genome by means of spatial autocorrelation analysis. This puts Galtier in a position to study variation in the strength of gBGC at three different scales, i) among species, ii) among genes, and iii) within genes. Galtier applies his method to four families of mammals, Hominidae, Cercopithecidae, Bovidae, and Muridae and provides a thorough discussion of his findings and methodology. Galtier found that the strength of gBGC correlates with proxies of the effective population size (Ne), but that the slope of the relationship differs among the four families of mammals. Given the relationship between the population-scaled strength of gBGC B = 4Neb, this finding suggests that the conversion bias (b) could vary among mammalian species. Variation in b could either result from differences in the strength of the transmission distortion (Galtier et al. 2018) or evolutionary changes in the rate of recombination (Boman et al. 2021). Alternatively, Galtier suggests that also systematic variation in proxies of Ne could lead to similar observations. Finally, the present study reports intriguing inter-species differences between the extent of variation in the strength of gBGC among and within genes, which are interpreted in consideration of the recombination dynamics in mammals. References Boman J, Mugal CF, Backström N (2021) The Effects of GC-Biased Gene Conversion on Patterns of Genetic Diversity among and across Butterfly Genomes. Genome Biology and Evolution, 13. https://doi.org/10.1093/gbe/evab064 Capra JA, Hubisz MJ, Kostka D, Pollard KS, Siepel A (2013) A Model-Based Analysis of GC-Biased Gene Conversion in the Human and Chimpanzee Genomes. PLOS Genetics, 9, e1003684. https://doi.org/10.1371/journal.pgen.1003684 De Maio N, Schlötterer C, Kosiol C (2013) Linking Great Apes Genome Evolution across Time Scales Using Polymorphism-Aware Phylogenetic Models. Molecular Biology and Evolution, 30, 2249–2262. https://doi.org/10.1093/molbev/mst131 Duret L, Galtier N (2009) Biased Gene Conversion and the Evolution of Mammalian Genomic Landscapes. Annual Review of Genomics and Human Genetics, 10, 285–311. https://doi.org/10.1146/annurev-genom-082908-150001 Eyre-Walker A (1999) Evidence of Selection on Silent Site Base Composition in Mammals: Potential Implications for the Evolution of Isochores and Junk DNA. Genetics, 152, 675–683. https://doi.org/10.1093/genetics/152.2.675 Galtier N (2021) Fine-scale quantification of GC-biased gene conversion intensity in mammals. bioRxiv, 2021.05.05.442789, ver. 5 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2021.05.05.442789 Galtier N, Piganeau G, Mouchiroud D, Duret L (2001) GC-Content Evolution in Mammalian Genomes: The Biased Gene Conversion Hypothesis. Genetics, 159, 907–911. https://doi.org/10.1093/genetics/159.2.907 Galtier N, Roux C, Rousselle M, Romiguier J, Figuet E, Glémin S, Bierne N, Duret L (2018) Codon Usage Bias in Animals: Disentangling the Effects of Natural Selection, Effective Population Size, and GC-Biased Gene Conversion. Molecular Biology and Evolution, 35, 1092–1103. https://doi.org/10.1093/molbev/msy015 Glémin S, Arndt PF, Messer PW, Petrov D, Galtier N, Duret L (2015) Quantification of GC-biased gene conversion in the human genome. Genome Research, 25, 1215–1228. https://doi.org/10.1101/gr.185488.114 Lartillot N (2013) Phylogenetic Patterns of GC-Biased Gene Conversion in Placental Mammals and the Evolutionary Dynamics of Recombination Landscapes. Molecular Biology and Evolution, 30, 489–502. https://doi.org/10.1093/molbev/mss239 Mugal CF, Weber CC, Ellegren H (2015) GC-biased gene conversion links the recombination landscape and demography to genomic base composition. BioEssays, 37, 1317–1326. https://doi.org/10.1002/bies.201500058 Nagylaki T (1983) Evolution of a finite population under gene conversion. Proceedings of the National Academy of Sciences, 80, 6278–6281. https://doi.org/10.1073/pnas.80.20.6278 | Fine-scale quantification of GC-biased gene conversion intensity in mammals | Nicolas Galtier | <p style="text-align: justify;">GC-biased gene conversion (gBGC) is a molecular evolutionary force that favours GC over AT alleles irrespective of their fitness effect. Quantifying the variation in time and across genomes of its intensity is key t... | | Evolutionary genomics, Population genomics, Vertebrates | Carina Farah Mugal | 2021-05-25 09:25:52 | ||
09 Aug 2023
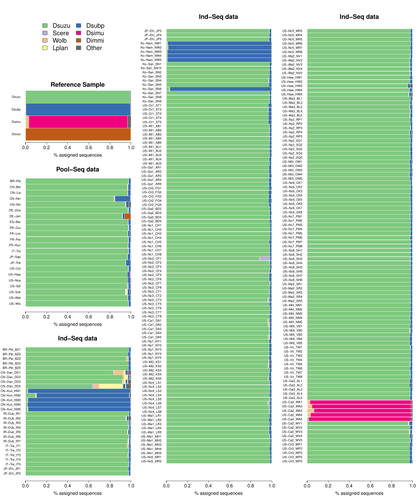
Efficient k-mer based curation of raw sequence data: application in Drosophila suzukiiDecontaminating reads, not contigsRecommended by Nicolas Galtier based on reviews by Marie Cariou and Denis BaurainContamination, the presence of foreign DNA sequences in a sample of interest, is currently a major problem in genomics. Because contamination is often unavoidable at the experimental stage, it is increasingly recognized that the processing of high-throughput sequencing data must include a decontamination step. This is usually performed after the many sequence reads have been assembled into a relatively small number of contigs. Dubious contigs are then discarded based on their composition (e.g. GC-content) or because they are highly similar to a known piece of DNA from a foreign species. Here [1], Mathieu Gautier explores a novel strategy consisting in decontaminating reads, not contigs. Why is this promising? Assembly programs and algorithms are complex, and it is not easy to predict, or monitor, how they handle contaminant reads. Ideally, contaminant reads will be assembled into obvious contaminant contigs. However, there might be more complex situations, such as chimeric contigs with alternating genuine and contaminant segments. Decontaminating at the read level, if possible, should eliminate such unfavorable situations where sequence information from contaminant and target samples are intimately intertwined by an assembler. To achieve this aim, Gautier proposes to use methods initially designed for the analysis of metagenomic data. This is pertinent since the decontamination process involves considering a sample as a mixture of different sources of DNA. The programs used here, CLARK and CLARK-L, are based on so-called k-mer analysis, meaning that the similarity between a read to annotate and a reference sequence is measured by how many sub-sequences (of length 31 base pairs for CLARK and 27 base pairs for CLARK-L) they share. This is notoriously more efficient than traditional sequence alignment algorithms when it comes to comparing a very large number of (most often unrelated) sequences. This is, therefore, a reference-based approach, in which the reads from a sample are assigned to previously sequenced genomes based on k-mer content. This original approach is here specifically applied to the case of Drosophila suzukii, an invasive pest damaging fruit production in Europe and America. Fortunately, Drosophila is a genus of insects with abundant genomic resources, including high-quality reference genomes in dozens of species. Having calibrated and validated his pipeline using data sets of known origins, Gautier quantifies in each of 258 presumed D. suzukii samples the proportion of reads that likely belong to other species of fruit flies, or to fruit fly-associated microbes. This proportion is close to one in 16 samples, which clearly correspond to mis-labelled individuals. It is non-negligible in another ~10 samples, which really correspond to D. suzukii individuals. Most of these reads of unexpected origin are contaminants and should be filtered out. Interestingly, one D. suzukii sample contains a substantial proportion of reads from the closely related D. subpulchera, which might instead reflect a recent episode of gene flow between these two species. The approach, therefore, not only serves as a crucial technical step, but also has the potential to reveal biological processes. Gautier's thorough, well-documented work will clearly benefit the ongoing and future research on D. suzuki, and Drosophila genomics in general. The author and reviewers rightfully note that, like any reference-based approach, this method is heavily dependent on the availability and quality of reference genomes - Drosophila being a favorable case. Building the reference database is a key step, and the interpretation of the output can only be made in the light of its content and gaps, as illustrated by Gautier's careful and detailed discussion of his numerous results. This pioneering study is a striking demonstration of the potential of metagenomic methods for the decontamination of high-throughput sequence data at the read level. The pipeline requires remarkably few computing resources, ensuring low carbon emission. I am looking forward to seeing it applied to a wide range of taxa and samples.
Reference [1] Gautier Mathieu. Efficient k-mer based curation of raw sequence data: application in Drosophila suzukii. bioRxiv, 2023.04.18.537389, ver. 2, peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2023.04.18.537389 | Efficient k-mer based curation of raw sequence data: application in *Drosophila suzukii* | Gautier Mathieu | <p>Several studies have highlighted the presence of contaminated entries in public sequence repositories, calling for special attention to the associated metadata. Here, we propose and evaluate a fast and efficient kmer-based approach to assess th... | | Bioinformatics, Population genomics | Nicolas Galtier | 2023-04-20 22:05:13 | ||
19 Jul 2021
TransPi - a comprehensive TRanscriptome ANalysiS PIpeline for de novo transcriptome assemblyTransPI: A balancing act between transcriptome assemblersRecommended by Oleg Simakov based on reviews by Gustavo Sanchez and Juan Daniel Montenegro CabreraEver since the introduction of the first widely usable assemblers for transcriptomic reads (Huang and Madan 1999; Schulz et al. 2012; Simpson et al. 2009; Trapnell et al. 2010, and many more), it has been a technical challenge to compare different methods and to choose the “right” or “best” assembly. It took years until the first widely accepted set of benchmarks beyond raw statistical evaluation became available (e.g., Parra, Bradnam, and Korf 2007; Simão et al. 2015). However, an approach to find the right balance between the number of transcripts or isoforms vs. evolutionary completeness measures has been lacking. This has been particularly pronounced in the field of non-model organisms (i.e., wild species that lack a genomic reference). Often, studies in this area employed only one set of assembly tools (the most often used to this day being Trinity, Haas et al. 2013; Grabherr et al. 2011). While it was relatively straightforward to obtain an initial assembly, its validation, annotation, as well its application to the particular purpose that the study was designed for (phylogenetics, differential gene expression, etc) lacked a clear workflow. This led to many studies using a custom set of tools with ensuing various degrees of reproducibility. TransPi (Rivera-Vicéns et al. 2021) fills this gap by first employing a meta approach using several available transcriptome assemblers and algorithms to produce a combined and reduced transcriptome assembly, then validating and annotating the resulting transcriptome. Notably, TransPI performs an extensive analysis/detection of chimeric transcripts, the results of which show that this new tool often produces fewer misassemblies compared to Trinity. TransPI not only generates a final report that includes the most important plots (in clickable/zoomable format) but also stores all relevant intermediate files, allowing advanced users to take a deeper look and/or experiment with different settings. As running TransPi is largely automated (including its installation via several popular package managers), it is very user-friendly and is likely to become the new "gold standard" for transcriptome analyses, especially of non-model organisms. References Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature Biotechnology, 29, 644–652. https://doi.org/10.1038/nbt.1883 Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, Couger MB, Eccles D, Li B, Lieber M, MacManes MD, Ott M, Orvis J, Pochet N, Strozzi F, Weeks N, Westerman R, William T, Dewey CN, Henschel R, LeDuc RD, Friedman N, Regev A (2013) De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nature Protocols, 8, 1494–1512. https://doi.org/10.1038/nprot.2013.084 Huang X, Madan A (1999) CAP3: A DNA Sequence Assembly Program. Genome Research, 9, 868–877. https://doi.org/10.1101/gr.9.9.868 Parra G, Bradnam K, Korf I (2007) CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics, 23, 1061–1067. https://doi.org/10.1093/bioinformatics/btm071 Rivera-Vicéns RE, Garcia-Escudero CA, Conci N, Eitel M, Wörheide G (2021) TransPi – a comprehensive TRanscriptome ANalysiS PIpeline for de novo transcriptome assembly. bioRxiv, 2021.02.18.431773, ver. 3 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2021.02.18.431773 Schulz MH, Zerbino DR, Vingron M, Birney E (2012) Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics, 28, 1086–1092. https://doi.org/10.1093/bioinformatics/bts094 Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM (2015) BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics, 31, 3210–3212. https://doi.org/10.1093/bioinformatics/btv351 Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJM, Birol İ (2009) ABySS: A parallel assembler for short read sequence data. Genome Research, 19, 1117–1123. https://doi.org/10.1101/gr.089532.108 Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L (2010) Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature Biotechnology, 28, 511–515. https://doi.org/10.1038/nbt.1621 | TransPi - a comprehensive TRanscriptome ANalysiS PIpeline for de novo transcriptome assembly | Ramon E Rivera-Vicens, Catalina Garcia-Escudero, Nicola Conci, Michael Eitel, Gert Wörheide | <p style="text-align: justify;">The use of RNA-Seq data and the generation of de novo transcriptome assemblies have been pivotal for studies in ecology and evolution. This is distinctly true for non-model organisms, where no genome information is ... | | Bioinformatics, Evolutionary genomics | Oleg Simakov | 2021-02-18 20:56:08 |




