
SIMAKOV Oleg
- Department of Molecular Evolution and Development, University of Vienna, Vienna, Austria
- Evolutionary genomics, Functional genomics
- recommender
Recommendation: 1
Reviews: 0
Recommendation: 1
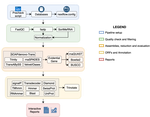
TransPi - a comprehensive TRanscriptome ANalysiS PIpeline for de novo transcriptome assembly
TransPI: A balancing act between transcriptome assemblers
Recommended by Oleg Simakov based on reviews by Gustavo Sanchez and Juan Daniel Montenegro CabreraEver since the introduction of the first widely usable assemblers for transcriptomic reads (Huang and Madan 1999; Schulz et al. 2012; Simpson et al. 2009; Trapnell et al. 2010, and many more), it has been a technical challenge to compare different methods and to choose the “right” or “best” assembly. It took years until the first widely accepted set of benchmarks beyond raw statistical evaluation became available (e.g., Parra, Bradnam, and Korf 2007; Simão et al. 2015). However, an approach to find the right balance between the number of transcripts or isoforms vs. evolutionary completeness measures has been lacking. This has been particularly pronounced in the field of non-model organisms (i.e., wild species that lack a genomic reference). Often, studies in this area employed only one set of assembly tools (the most often used to this day being Trinity, Haas et al. 2013; Grabherr et al. 2011). While it was relatively straightforward to obtain an initial assembly, its validation, annotation, as well its application to the particular purpose that the study was designed for (phylogenetics, differential gene expression, etc) lacked a clear workflow. This led to many studies using a custom set of tools with ensuing various degrees of reproducibility.
TransPi (Rivera-Vicéns et al. 2021) fills this gap by first employing a meta approach using several available transcriptome assemblers and algorithms to produce a combined and reduced transcriptome assembly, then validating and annotating the resulting transcriptome. Notably, TransPI performs an extensive analysis/detection of chimeric transcripts, the results of which show that this new tool often produces fewer misassemblies compared to Trinity. TransPI not only generates a final report that includes the most important plots (in clickable/zoomable format) but also stores all relevant intermediate files, allowing advanced users to take a deeper look and/or experiment with different settings. As running TransPi is largely automated (including its installation via several popular package managers), it is very user-friendly and is likely to become the new "gold standard" for transcriptome analyses, especially of non-model organisms.
References
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature Biotechnology, 29, 644–652. https://doi.org/10.1038/nbt.1883
Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, Couger MB, Eccles D, Li B, Lieber M, MacManes MD, Ott M, Orvis J, Pochet N, Strozzi F, Weeks N, Westerman R, William T, Dewey CN, Henschel R, LeDuc RD, Friedman N, Regev A (2013) De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nature Protocols, 8, 1494–1512. https://doi.org/10.1038/nprot.2013.084
Huang X, Madan A (1999) CAP3: A DNA Sequence Assembly Program. Genome Research, 9, 868–877. https://doi.org/10.1101/gr.9.9.868
Parra G, Bradnam K, Korf I (2007) CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics, 23, 1061–1067. https://doi.org/10.1093/bioinformatics/btm071
Rivera-Vicéns RE, Garcia-Escudero CA, Conci N, Eitel M, Wörheide G (2021) TransPi – a comprehensive TRanscriptome ANalysiS PIpeline for de novo transcriptome assembly. bioRxiv, 2021.02.18.431773, ver. 3 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2021.02.18.431773
Schulz MH, Zerbino DR, Vingron M, Birney E (2012) Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics, 28, 1086–1092. https://doi.org/10.1093/bioinformatics/bts094
Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM (2015) BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics, 31, 3210–3212. https://doi.org/10.1093/bioinformatics/btv351
Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJM, Birol İ (2009) ABySS: A parallel assembler for short read sequence data. Genome Research, 19, 1117–1123. https://doi.org/10.1101/gr.089532.108
Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L (2010) Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature Biotechnology, 28, 511–515. https://doi.org/10.1038/nbt.1621