
Latest recommendations

| Id | Title | Authors | Abstract▼ | Picture | Thematic fields | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
22 Nov 2023
The slow evolving genome of the xenacoelomorph worm Xenoturbella bockiGenomic idiosyncrasies of Xenoturbella bocki: morphologically simple yet genetically complexRecommended by Rosa Fernández based on reviews by Christopher Laumer and 1 anonymous reviewerXenoturbella is a genus of morphologically simple bilaterians inhabiting benthic environments. Until very recently, only one species was known from the genus, Xenoturbella bocki Westblad 1949 [1]. Less than a decade ago, five more species were discovered (X. churro, X. monstrosa, X. profunda, X. hollandorum [2] and X. japonica [3]). These enigmatic animals lack an anus, a coelom, reproductive organs, nephrocytes and a centralized nervous system [1]. The systematic classification of the genus has substantially changed in the last decades, with first being considered as its own phylum (Xenoturbellida) and then being clustered together with acoels and nemertodermatids into the phylum Xenacoelomorpha [4,5]. The phylogenetic position of the xenacoelomorphs has been recalcitrant to resolution, with its position ranging from being the sister group to Nephrozoa (ie, protostomes and deuterostomes [6]) to the sister group to Ambulacraria (ie, Hemichordata and Echinodermata) in a clade called Xenambulacraria [4]. Recent studies based on expanded datasets and more refined analyses support either topology [7,8]. Either way, it is clear that additional studies on Xenoturbella could provide important insights into the origins of bilaterian traits such as the anus, the nephrons and the evolution of a centralized nervous system.
In any case, we are approaching a qualitative jump in how we understand phylogenomics thanks to efforts derived from the availability of chromosome-level genome assemblies for a growing number of species. Exciting times are ahead for us, evolutionary biologists, to explore what high-quality genomes - in combination with multiomics datasets - will reveal about animal evolution. I am personally really looking forward to it. References 1. Westblad E. (1949). Xenoturbella bocki n.g., n.sp., a peculiar, primitive Turbellarian type. Arkiv för Zoologi 1, 3-29 (1949). 2. Rouse, G. W., Wilson, N. G., Carvajal, J. I. & Vrijenhoek, R. C. New deep-sea species of Xenoturbella and the position of Xenacoelomorpha. Nature 530, 94–97 (2016). https://doi.org/10.1038/nature16545 3. Nakano, H. et al. Correction to: A new species of Xenoturbella from the western Pacific Ocean and the evolution of Xenoturbella. BMC Evol. Biol. 18, 1–2 (2018). https://doi.org/10.1186/s12862-018-1190-5https://doi.org/10.1186/s12862-018-1190-5 4. Philippe, H. et al. Acoelomorph flatworms are deuterostomes related to Xenoturbella. Nature 470, 255–258 (2011). https://doi.org/10.1038/nature09676 5. Hejnol, A. et al. Assessing the root of bilaterian animals with scalable phylogenomic methods. Proc. Biol. Sci. 276, 4261–4270 (2009). https://doi.org/10.1098/rspb.2009.0896 6. Cannon, J. T. et al. Xenacoelomorpha is the sister group to Nephrozoa. Nature 530, 89–93 (2016). https://doi.org/10.1038/nature16520 7. Laumer, C. E. et al. Revisiting metazoan phylogeny with genomic sampling of all phyla. Proc. Biol. Sci. 286, 20190831 (2019). https://doi.org/10.1098/rspb.2019.0831 8. Philippe, H. et al. Mitigating anticipated effects of systematic errors supports sister-group relationship between Xenacoelomorpha and Ambulacraria. Curr. Biol. 29, 1818–1826.e6 (2019). https://doi.org/10.1016/j.cub.2019.04.009 9. Schiffer, P. H., Natsidis, P., Leite D. J., Robertson, H., Lapraz, F., Marlétaz, F., Fromm, B., Baudry, L., Simpson, F., Høye, E., Zakrzewski, A-C., Kapli, P., Hoff, K. J., Mueller, S., Marbouty, M., Marlow, H., Copley, R. R., Koszul, R., Sarkies, P. & Telford, M .J. The slow evolving genome of the xenacoelomorph worm Xenoturbella bocki. bioRxiv (2023), ver. 4 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2022.06.24.497508 10. Suga, H. et al. The Capsaspora genome reveals a complex unicellular prehistory of animals. Nat. Commun. 4, 2325 (2013). https://doi.org/10.1038/ncomms3325 11. Fernández, R. & Gabaldón, T. Gene gain and loss across the metazoan tree of life. Nat Ecol Evol 4, 524–533 (2020). https://doi.org/10.1038/s41559-019-1069-x | The slow evolving genome of the xenacoelomorph worm *Xenoturbella bocki* | Philipp H. Schiffer, Paschalis Natsidis, Daniel J. Leite, Helen Robertson, François Lapraz, Ferdinand Marlétaz, Bastian Fromm, Liam Baudry, Fraser Simpson, Eirik Høye, Anne-C. Zakrzewski, Paschalia Kapli, Katharina J. Hoff, Steven Mueller, Martial... | <p style="text-align: justify;">The evolutionary origins of Bilateria remain enigmatic. One of the more enduring proposals highlights similarities between a cnidarian-like planula larva and simple acoel-like flatworms. This idea is based in part o... | | Evolutionary genomics | Rosa Fernández | 2022-11-01 12:31:53 | ||
20 Jul 2021
Genetic mapping of sex and self-incompatibility determinants in the androdioecious plant Phillyrea angustifoliaIdentification of distinct YX-like loci for sex determination and self-incompatibility in an androdioecious shrubRecommended by Tatiana Giraud and Ricardo C. Rodríguez de la Vega based on reviews by 2 anonymous reviewersA wide variety of systems have evolved to control mating compatibility in sexual organisms. Their genetic determinism and the factors controlling their evolution represent fascinating questions in evolutionary biology and genomics. The plant Phillyrea angustifolia (Oleaeceae family) represents an exciting model organism, as it displays two distinct and rare mating compatibility systems [1]: 1) males and hermaphrodites co-occur in populations of this shrub (a rare system called androdioecy), while the evolution and maintenance of purely hermaphroditic plants or mixtures of females and hermaphrodites (a system called gynodioecy) are easier to explain [2]; 2) a homomorphic diallelic self-incompatibility system acts in hermaphrodites, while such systems are usually multi-allelic, as rare alleles are advantageous, being compatible with all other alleles. Previous analyses of crosses brought some interesting answers to these puzzles, showing that males benefit from the ability to mate with all hermaphrodites regardless of their allele at the self-incompatibility system, and suggesting that both sex and self incompatibility are determined by XY-like genetic systems, i.e. with each a dominant allele; homozygotes for a single allele and heterozygotes therefore co-occur in natural populations at both sex and self-incompatibility loci [3]. Here, Carré et al. used genotyping-by-sequencing to build a genome linkage map of P. angustifolia [4]. The elegant and original use of a probabilistic model of segregating alleles (implemented in the SEX-DETector method) allowed to identify both the sex and self-incompatibility loci [4], while this tool was initially developed for detecting sex-linked genes in species with strictly separated sexes (dioecy) [5]. Carré et al. [4] confirmed that the sex and self-incompatibility loci are located in two distinct linkage groups and correspond to XY-like systems. A comparison with the genome of the closely related Olive tree indicated that their self-incompatibility systems were homologous. Such a XY-like system represents a rare genetic determination mechanism for self-incompatibility and has also been recently found to control mating types in oomycetes [6]. This study [4] paves the way for identifying the genes controlling the sex and self-incompatibility phenotypes and for understanding why and how self-incompatibility is only expressed in hermaphrodites and not in males. It will also be fascinating to study more finely the degree and extent of genomic differentiation at these two loci and to assess whether recombination suppression has extended stepwise away from the sex and self-incompatibility loci, as can be expected under some hypotheses, such as the sheltering of deleterious alleles near permanently heterozygous alleles [7]. Furthermore, the co-occurrence in P. angustifolia of sex and mating types can contribute to our understanding of the factor controlling their evolution [8]. References [1] Saumitou-Laprade P, Vernet P, Vassiliadis C, Hoareau Y, Magny G de, Dommée B, Lepart J (2010) A Self-Incompatibility System Explains High Male Frequencies in an Androdioecious Plant. Science, 327, 1648–1650. https://doi.org/10.1126/science.1186687 [2] Pannell JR, Voillemot M (2015) Plant Mating Systems: Female Sterility in the Driver’s Seat. Current Biology, 25, R511–R514. https://doi.org/10.1016/j.cub.2015.04.044 [3] Billiard S, Husse L, Lepercq P, Godé C, Bourceaux A, Lepart J, Vernet P, Saumitou-Laprade P (2015) Selfish male-determining element favors the transition from hermaphroditism to androdioecy. Evolution, 69, 683–693. https://doi.org/10.1111/evo.12613 [4] Carre A, Gallina S, Santoni S, Vernet P, Gode C, Castric V, Saumitou-Laprade P (2021) Genetic mapping of sex and self-incompatibility determinants in the androdioecious plant Phillyrea angustifolia. bioRxiv, 2021.04.15.439943, ver. 7 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2021.04.15.439943 [5] Muyle A, Käfer J, Zemp N, Mousset S, Picard F, Marais GA (2016) SEX-DETector: A Probabilistic Approach to Study Sex Chromosomes in Non-Model Organisms. Genome Biology and Evolution, 8, 2530–2543. https://doi.org/10.1093/gbe/evw172 [6] Dussert Y, Legrand L, Mazet ID, Couture C, Piron M-C, Serre R-F, Bouchez O, Mestre P, Toffolatti SL, Giraud T, Delmotte F (2020) Identification of the First Oomycete Mating-type Locus Sequence in the Grapevine Downy Mildew Pathogen, Plasmopara viticola. Current Biology, 30, 3897-3907.e4. https://doi.org/10.1016/j.cub.2020.07.057 [7] Jay P, Tezenas E, Giraud T (2021) A deleterious mutation-sheltering theory for the evolution of sex chromosomes and supergenes. bioRxiv, 2021.05.17.444504. https://doi.org/10.1101/2021.05.17.444504 [8] Billiard S, López-Villavicencio M, Devier B, Hood ME, Fairhead C, Giraud T (2011) Having sex, yes, but with whom? Inferences from fungi on the evolution of anisogamy and mating types. Biological Reviews, 86, 421–442. https://doi.org/10.1111/j.1469-185X.2010.00153.x | Genetic mapping of sex and self-incompatibility determinants in the androdioecious plant Phillyrea angustifolia | Amelie Carre, Sophie Gallina, Sylvain Santoni, Philippe Vernet, Cecile Gode, Vincent Castric, Pierre Saumitou-Laprade | <p style="text-align: justify;">The diversity of mating and sexual systems in angiosperms is spectacular, but the factors driving their evolution remain poorly understood. In plants of the Oleaceae family, an unusual self-incompatibility (SI) syst... | | Evolutionary genomics, Plants | Tatiana Giraud | 2021-05-04 10:37:26 | ||
20 Nov 2023
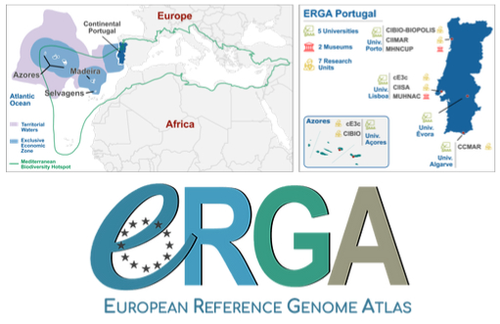
Building a Portuguese Coalition for Biodiversity GenomicsThe Portuguese genomics community teams up with iconic species to understand the destruction of biodiversityRecommended by Fernando Racimo based on reviews by Svein-Ole Mikalsen and 1 anonymous reviewerThis manuscript describes the ongoing work and plans of Biogenome Portugal: a new network of researchers in the Portuguese biodiversity genomics community. The aims of this network are to jointly train scientists in ecology and evolution, generate new knowledge and understanding of Portuguese biodiversity, and better engage with the public and with international researchers, so as to advance conservation efforts in the region. In collaboration across disciplines and institutions, they are also contributing to the European Reference Genome Atlas (ERGA): a massive scientific effort, seeking to eventually produce reference-quality genomes for all species in the European continent (Mc Cartney et al. 2023). The manuscript centers around six iconic and/or severely threatened species, whose range extends across parts of what is today considered Portuguese territory. Via the Portugal chapter of ERGA (ERGA-Portugal), the researchers will generate high-quality genome sequences from these species. The species are the Iberian hare, the Azores laurel, the Black wheatear, the Portuguese crowberry, the Cave ground beetle and the Iberian minnowcarp. In ignorance of human-made political borders, some of these species also occupy large parts of the rest of the Iberian peninsula, highlighting the importance of transnational collaboration in biodiversity efforts. The researchers extracted samples from members of each of these species, and are building reference genome sequences from them. In some cases, these sequences will also be co-analyzed with additional population genomic data from the same species or genetic data from cohabiting species. The researchers aim to answer a variety of ecological and evolutionary questions using this information, including how genetic diversity is being affected by the destruction of their habitat, and how they are being forced to adapt as a consequence of the climate emergency. The authors did a very good job in providing a justification for the choice of pilot species, a thorough methodological overview of current work, and well thought-out plans for future analyses once the genome sequences are available for study. The authors also describe plans for networking and training activities to foster a well-connected Portuguese biodiversity genomics community. Applying a genomic analysis lens is important for understanding the ever faster process of devastation of our natural world. Governments and corporations around the globe are destroying nature at ever larger scales (Diaz et al. 2019). They are also destabilizing the climatic conditions on which life has existed for thousands of years (Trisos et al. 2020). Thus, genetic diversity is decreasing faster than ever in human history, even when it comes to non-threatened species (Exposito-Alonso et al. 2022), and these decreases are disrupting ecological processes worldwide (Richardson et al. 2023). This, in turn, is threatening the conditions on which the stability of our societies rest (Gardner and Bullock 2021). The efforts of Biogenome Portal and ERGA-Portugal will go a long way in helping us understand in greater detail how this process is unfolding in Portuguese territories.
References Díaz, Sandra, et al. "Pervasive human-driven decline of life on Earth points to the need for transformative change." Science 366.6471 (2019): eaax3100. https://doi.org/10.1126/science.aax3100 Exposito-Alonso, Moises, et al. "Genetic diversity loss in the Anthropocene." Science 377.6613 (2022): 1431-1435. https://doi.org/10.1126/science.abn5642 Gardner, Charlie J., and James M. Bullock. "In the climate emergency, conservation must become survival ecology." Frontiers in Conservation Science 2 (2021): 659912. https://doi.org/10.3389/fcosc.2021.659912 Mc Cartney, Ann M., et al. "The European Reference Genome Atlas: piloting a decentralised approach to equitable biodiversity genomics." bioRxiv (2023): 2023-09, ver. 2 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.32942/X20W3Q Richardson, Katherine, et al. "Earth beyond six of nine planetary boundaries." Science Advances 9.37 (2023): eadh2458. https://doi.org/10.1126/sciadv.adh2458 Trisos, Christopher H., Cory Merow, and Alex L. Pigot. "The projected timing of abrupt ecological disruption from climate change." Nature 580.7804 (2020): 496-501. https://doi.org/10.1038/s41586-020-2189-9 | Building a Portuguese Coalition for Biodiversity Genomics | João Pedro Marques, Paulo Célio Alves, Isabel R. Amorim, Ricardo J. Lopes, Mónica Moura, Gene Meyers, Manuela Sim-Sim, Carla Sousa-Santos, Maria Judite Alves, Paulo AV Borges, Thomas Brown, Miguel Carneiro, Carlos Carrapato, Luís Ceríaco, Claudio ... | <p style="text-align: justify;">The diverse physiography of the Portuguese land and marine territory, spanning from continental Europe to the Atlantic archipelagos, has made it an important repository of biodiversity throughout the Pleistocene gla... | | ERGA, ERGA Pilot | Fernando Racimo | 2023-07-14 11:24:22 | ||
25 Nov 2022
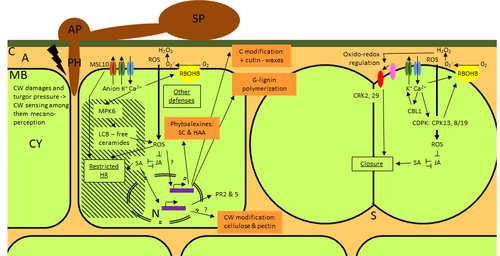
Phenotypic and transcriptomic analyses reveal major differences between apple and pear scab nonhost resistanceApples and pears: two closely related species with differences in scab nonhost resistanceRecommended by Wirulda Pootakham based on reviews by 3 anonymous reviewersNonhost resistance is a common form of disease resistance exhibited by plants against microorganisms that are pathogenic to other plant species [1]. Apples and pears are two closely related species belonging to Rosaceae family, both affected by scab disease caused by fungal pathogens in the Venturia genus. These pathogens appear to be highly host-specific. While apples are nonhosts for Venturia pyrina, pears are nonhosts for Venturia inaequalis. To date, the molecular bases of scab nonhost resistance in apple and pear have not been elucidated. This preprint by Vergne, et al (2022) [2] analyzed nonhost resistance symptoms in apple/V. pyrina and pear/V. inaequalis interactions as well as their transcriptomic responses. Interestingly, the author demonstrated that the nonhost apple/V. pyrina interaction was almost symptomless while hypersensitive reactions were observed for pear/V. inaequalis interaction. The transcriptomic analyses also revealed a number of differentially expressed genes (DEGs) that corresponded to the severity of the interactions, with very few DEGs observed during the apple/V. pyrina interaction and a much higher number of DEGs during the pear/V. inaequalis interaction. This type of reciprocal host-pathogen interaction study is valuable in gaining new insights into how plants interact with microorganisms that are potential pathogens in related species. A few processes appeared to be involved in the pear resistance against the nonhost pathogen V. inaequalis at the transcriptomic level, such as stomata closure, modification of cell wall and production of secondary metabolites as well as phenylpropanoids. Based on the transcriptomics changes during the nonhost interaction, the author compared the responses to those of host-pathogen interactions and revealed some interesting findings. They proposed a series of cascading effects in pear induced by the presence of V. inaequalis, which I believe helps shed some light on the basic mechanism for nonhost resistance. I am recommending this study because it provides valuable information that will strengthen our understanding of nonhost resistance in the Rosaceae family and other plant species. The knowledge gained here may be applied to genetically engineer plants for a broader resistance against a number of pathogens in the future. References 1. Senthil-Kumar M, Mysore KS (2013) Nonhost Resistance Against Bacterial Pathogens: Retrospectives and Prospects. Annual Review of Phytopathology, 51, 407–427. https://doi.org/10.1146/annurev-phyto-082712-102319 2. Vergne E, Chevreau E, Ravon E, Gaillard S, Pelletier S, Bahut M, Perchepied L (2022) Phenotypic and transcriptomic analyses reveal major differences between apple and pear scab nonhost resistance. bioRxiv, 2021.06.01.446506, ver. 4 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2021.06.01.446506 | Phenotypic and transcriptomic analyses reveal major differences between apple and pear scab nonhost resistance | E. Vergne, E. Chevreau, E. Ravon, S. Gaillard, S. Pelletier, M. Bahut, L. Perchepied | <p style="text-align: justify;"><strong>Background. </strong>Nonhost resistance is the outcome of most plant/pathogen interactions, but it has rarely been described in Rosaceous fruit species. Apple (<em>Malus x domestica</em> Borkh.) have a nonho... | | Functional genomics, Plants | Wirulda Pootakham | Jessica Soyer, Anonymous | 2022-05-13 15:06:08 | |
10 Jul 2023
SNP discovery by exome capture and resequencing in a pea genetic resource collectionThe value of a large Pisum SNP datasetRecommended by Wanapinun Nawae based on reviews by Rui Borges and 1 anonymous reviewerOne important goal of modern genetics is to establish functional associations between genotype and phenotype. Single nucleotide polymorphisms (SNPs) are numerous and widely distributed in the genome and can be obtained from nucleic acid sequencing (1). SNPs allow for the investigation of genetic diversity, which is critical for increasing crop resilience to the challenges posed by global climate change. The associations between SNPs and phenotypes can be captured in genome-wide association studies. SNPs can also be used in combination with machine learning, which is becoming more popular for predicting complex phenotypic traits like yield and biotic and abiotic stress tolerance from genotypic data (2). The availability of many SNP datasets is important in machine learning predictions because this approach requires big data to build a comprehensive model of the association between genotype and phenotype. Aubert and colleagues have studied, as part of the PeaMUST project, the genetic diversity of 240 Pisum accessions (3). They sequenced exome-enriched genomic libraries, a technique that enables the identification of high-density, high-quality SNPs at a low cost (4). This technique involves capturing and sequencing only the exonic regions of the genome, which are the protein-coding regions. A total of 2,285,342 SNPs were obtained in this study. The analysis of these SNPs with the annotations of the genome sequence of one of the studied pea accessions (5) identified a number of SNPs that could have an impact on gene activity. Additional analyses revealed 647,220 SNPs that were unique to individual pea accessions, which might contribute to the fitness and diversity of accessions in different habitats. Phylogenetic and clustering analyses demonstrated that the SNPs could distinguish Pisum germplasms based on their agronomic and evolutionary histories. These results point out the power of selected SNPs as markers for identifying Pisum individuals. Overall, this study found high-quality SNPs that are meaningful in a biological context. This dataset was derived from a large set of germplasm and is thus particularly useful for studying genotype-phenotype associations, as well as the diversity within Pisum species. These SNPs could also be used in breeding programs to develop new pea varieties that are resilient to abiotic and biotic stressors. References
https://doi.org/10.1139/gen-2021-005
https://doi.org/10.1186/s12870-022-03559-z
https://doi.org/10.1101/2022.08.03.502586
https://doi.org/10.1534/g3.115.018564
| SNP discovery by exome capture and resequencing in a pea genetic resource collection | G. Aubert, J. Kreplak, M. Leveugle, H. Duborjal, A. Klein, K. Boucherot, E. Vieille, M. Chabert-Martinello, C. Cruaud, V. Bourion, I. Lejeune-Hénaut, M.L. Pilet-Nayel, Y. Bouchenak-Khelladi, N. Francillonne, N. Tayeh, J.P. Pichon, N. Rivière, J. B... | <p style="text-align: justify;"><strong>Background & Summary</strong></p> <p style="text-align: justify;">In addition to being the model plant used by Mendel to establish genetic laws, pea (<em>Pisum sativum</em> L., 2n=14) is a major pulse c... | | Plants, Population genomics | Wanapinun Nawae | 2022-11-29 09:29:06 | ||
22 May 2023

Genetic bases of resistance to the rice hoja blanca disease deciphered by a QTL approachScoring symptoms of a plant viral diseaseRecommended by Olivier Panaud based on reviews by Grégoire Aubert and Valérie GeffroyThe paper from Silva et al. (2023) provides new insights into the genetic bases of natural resistance of rice to the Rice Hoja Blanca (RHB) disease, one of its most serious diseases in tropical countries of the American continent and the Caribbean. This disease is caused by the Rice Hoja Blanca Virus, or RHBV, the vector of which is the planthopper insect Tagosodes orizicolus Müir. It is responsible for serious damage to the rice crop (Morales and Jennings 2010). The authors take a Quantitative Trait Loci (QTL) detection approach to find genomic regions statistically associated with the resistant phenotype. To this aim, they use four resistant x susceptible crosses (the susceptible parent being the same in all four crosses) to maximize the chances to find new QTLs. The F2 populations derived from the crosses are genotyped using Single Nucleotide Polymorphisms (SNPs) extracted from whole-genome sequencing (WGS) data of the resistant parents, and the F3 families derived from the F2 individuals are scored for disease symptoms. For this, they use a computer-aided image analysis protocol that they designed so they can estimate the severity of the damages in the plant. They find several new QTLs, some being apparently more associated with disease severity, others with disease incidence. They also find that a previously identified QTL of Oryza sativa ssp. japonica origin is also present in the indica cluster (Romero et al. 2014). Finally, they discuss the candidate genes that could underlie the QTLs and provide a simple model for resistance. It has to be noted that scoring symptoms of a viral disease such as RHB is very challenging. It requires maintaining populations of viruliferous insect vectors, mastering times and conditions for infestation by nymphs, and precise symptom scoring. It also requires the preparation of segregating populations, their genotyping with enough genetic markers, and mastering QTL detection methods. All these aspects are present in this work. In particular, the phenotyping of symptom severity implemented using computer-aided image processing represents an impressive, enormous amount of work. From the genomics side, the fine-scale genotyping is based on the WGS of the parental lines (resistant and susceptible), followed by the application of suitable bioinformatic tools for SNP extraction and primers prediction that can be used on their Fluidigm platform. It also required implementing data correction algorithms to achieve precise genetic maps in the four crosses. The QTL detection itself required careful statistical pre-processing of phenotypic data. The authors then used a combination of several QTL detection methods, including an original meta-QTL method they developed in the software MapDisto. The authors then perform a very complete and convincing analysis of candidate genes, which includes genes already identified for a similar disease (RSV) on chromosome 11 of rice. What remains to elucidate is whether the candidate genes are actually involved or not in the disease resistance process. The team has already started implementing gene knockout strategies to study some of them in more detail. It will be interesting to see whether those genes act against the virus itself, or against the insect vector. Overall the work is of high quality and represents an important advance in the knowledge of disease resistance. In addition, it has many implications for crop breeding, allowing the setup of large-scale, marker-assisted strategies, for new resistant elite varieties of rice. References Morales F and Jennings P (2010) Rice hoja blanca: a complex plant-virus-vector pathosystem. CAB Reviews. https://doi.org/10.1079/PAVSNNR20105043 Romero LE, Lozano I, Garavito A, et al (2014) Major QTLs control resistance to Rice hoja blanca virus and its vector Tagosodes orizicolus. G3 | Genes, Genomes, Genetics 4:133–142. https://doi.org/10.1534/g3.113.009373 Silva A, Montoya ME, Quintero C, Cuasquer J, Tohme J, Graterol E, Cruz M, Lorieux M (2023) Genetic bases of resistance to the rice hoja blanca disease deciphered by a QTL approach. bioRxiv, 2022.11.07.515427, ver. 2 peer-reviewed and recommended by Peer Community in Genomics https://doi.org/10.1101/2022.11.07.515427 | Genetic bases of resistance to the rice hoja blanca disease deciphered by a QTL approach | Alexander Silva, Maria Elker Montoya, Constanza Quintero, Juan Cuasquer, Joe Tohme, Eduardo Graterol, Maribel Cruz, Mathias Lorieux | <p style="text-align: justify;">Rice hoja blanca (RHB) is one of the most serious diseases in rice growing areas in tropical Americas. Its causal agent is Rice hoja blanca virus (RHBV), transmitted by the planthopper <em>Tagosodes orizicolus </em>... | | Functional genomics, Plants | Olivier Panaud | 2022-11-09 09:13:30 | ||
11 Sep 2023
COVFlow: phylodynamics analyses of viruses from selected SARS-CoV-2 genome sequencesA pipeline to select SARS-CoV-2 sequences for reliable phylodynamic analysesRecommended by Emmanuelle Lerat based on reviews by Gabriel Wallau and Bastien BoussauPhylodynamic approaches enable viral genetic variation to be tracked over time, providing insight into pathogen phylogenetic relationships and epidemiological dynamics. These are important methods for monitoring viral spread, and identifying important parameters such as transmission rate, geographic origin and duration of infection [1]. This knowledge makes it possible to adjust public health measures in real-time and was important in the case of the COVID-19 pandemic [2]. However, these approaches can be complicated to use when combining a very large number of sequences. This was particularly true during the COVID-19 pandemic, when sequencing data representing millions of entire viral genomes was generated, with associated metadata enabling their precise identification. Danesh et al. [3] present a bioinformatics pipeline, CovFlow, for selecting relevant sequences according to user-defined criteria to produce files that can be used directly for phylodynamic analyses. The selection of sequences first involves a quality filter on the size of the sequences and the absence of unresolved bases before being able to make choices based on the associated metadata. Once the sequences are selected, they are aligned and a time-scaled phylogenetic tree is inferred. An output file in a format directly usable by BEAST 2 [4] is finally generated. To illustrate the use of the pipeline, Danesh et al. [3] present an analysis of the Delta variant in two regions of France. They observed a delay in the start of the epidemic depending on the region. In addition, they identified genetic variation linked to the start of the school year and the extension of vaccination, as well as the arrival of a new variant. This tool will be of major interest to researchers analysing SARS-CoV-2 sequencing data, and a number of future developments are planned by the authors. References [1] Baele G, Dellicour S, Suchard MA, Lemey P, Vrancken B. 2018. Recent advances in computational phylodynamics. Curr Opin Virol. 31:24-32. https://doi.org/10.1016/j.coviro.2018.08.009 [2] Attwood SW, Hill SC, Aanensen DM, Connor TR, Pybus OG. 2022. Phylogenetic and phylodynamic approaches to understanding and combating the early SARS-CoV-2 pandemic. Nat Rev Genet. 23:547-562. https://doi.org/10.1038/s41576-022-00483-8 [3] Danesh G, Boennec C, Verdurme L, Roussel M, Trombert-Paolantoni S, Visseaux B, Haim-Boukobza S, Alizon S. 2023. COVFlow: phylodynamics analyses of viruses from selected SARS-CoV-2 genome sequences. bioRxiv, ver. 7 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2022.06.17.496544 [4] Bouckaert R, Heled J, Kühnert D, Vaughan T, Wu C-H et al. 2014. BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput Biol 10: e1003537. https://doi.org/10.1371/journal.pcbi.1003537 | COVFlow: phylodynamics analyses of viruses from selected SARS-CoV-2 genome sequences | Gonché Danesh, Corentin Boennec, Laura Verdurme, Mathilde Roussel, Sabine Trombert-Paolantoni, Benoit Visseaux, Stephanie Haim-Boukobza, Samuel Alizon | <p style="text-align: justify;">Phylodynamic analyses generate important and timely data to optimise public health response to SARS-CoV-2 outbreaks and epidemics. However, their implementation is hampered by the massive amount of sequence data and... | | Bioinformatics, Evolutionary genomics | Emmanuelle Lerat | 2022-12-12 09:04:01 | ||
06 May 2022

A deep dive into genome assemblies of non-vertebrate animalsDiving, and even digging, into the wild jungle of annotation pathways for non-vertebrate animalsRecommended by Francois Sabot based on reviews by Yann Bourgeois, Cécile Monat, Valentina Peona and Benjamin Istace based on reviews by Yann Bourgeois, Cécile Monat, Valentina Peona and Benjamin Istace
In their paper, Guiglielmoni et al. propose we pick up our snorkels and palms and take "A deep dive into genome assemblies of non-vertebrate animals" (1). Indeed, while numerous assembly-related tools were developed and tested for human genomes (or at least vertebrates such as mice), very few were tested on non-vertebrate animals so far. Moreover, most of the benchmarks are aimed at raw assembly tools, and very few offer a guide from raw reads to an almost finished assembly, including quality control and phasing. This huge and exhaustive review starts with an overview of the current sequencing technologies, followed by the theory of the different approaches for assembly and their implementation. For each approach, the authors present some of the most representative tools, as well as the limits of the approach. The authors additionally present all the steps required to obtain an almost complete assembly at a chromosome-scale, with all the different technologies currently available for scaffolding, QC, and phasing, and the way these tools can be applied to non-vertebrates animals. Finally, they propose some useful advice on the choice of the different approaches (but not always tools, see below), and advocate for a robust genome database with all information on the way the assembly was obtained. This review is a very complete one for now and is a very good starting point for any student or scientist interested to start working on genome assembly, from either model or non-model organisms. However, the authors do not provide a list of tools or a benchmark of them as a recommendation. Why? Because such a proposal may be obsolete in less than a year.... Indeed, with the explosion of the 3rd generation of sequencing technology, assembly tools (from different steps) are constantly evolving, and their relative performance increases on a monthly basis. In addition, some tools are really efficient at the time of a review or of an article, but are not further developed later on, and thus will not evolve with the technology. We have all seen it with wonderful tools such as Chiron (2) or TopHat (3), which were very promising ones, but cannot be developed further due to the stop of the project, the end of the contract of the post-doc in charge of the development, or the decision of the developer to switch to another paradigm. Such advice would, therefore, need to be constantly updated. Thus, the manuscript from Guiglielmoni et al will be an almost intemporal one (up to the next sequencing revolution at last), and as they advocated for a more informed genome database, I think we should consider a rolling benchmarking system (tools, genome and sequence dataset) allowing to keep the performance of the tools up-to-date, and to propose the best set of assembly tools for a given type of genome. References 1. Guiglielmoni N, Rivera-Vicéns R, Koszul R, Flot J-F (2022) A Deep Dive into Genome Assemblies of Non-vertebrate Animals. Preprints, 2021110170, ver. 3 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.20944/preprints202111.0170 2. Teng H, Cao MD, Hall MB, Duarte T, Wang S, Coin LJM (2018) Chiron: translating nanopore raw signal directly into nucleotide sequence using deep learning. GigaScience, 7, giy037. https://doi.org/10.1093/gigascience/giy037 3. Trapnell C, Pachter L, Salzberg SL (2009) TopHat: discovering splice junctions with RNA-Seq. Bioinformatics, 25, 1105–1111. https://doi.org/10.1093/bioinformatics/btp120 | A deep dive into genome assemblies of non-vertebrate animals | Nadège Guiglielmoni, Ramón Rivera-Vicéns, Romain Koszul, Jean-François Flot | <p style="text-align: justify;">Non-vertebrate species represent about ∼95% of known metazoan (animal) diversity. They remain to this day relatively unexplored genetically, but understanding their genome structure and function is pivotal for expan... | | Bioinformatics, Evolutionary genomics | Francois Sabot | Valentina Peona, Benjamin Istace, Cécile Monat, Yann Bourgeois | 2021-11-10 17:47:31 | |
08 Apr 2022

POSTPRINT
Phylogenetics in the Genomic Era“Phylogenetics in the Genomic Era” brings together experts in the field to present a comprehensive synthesisRecommended by Robert Waterhouse and Karen MeusemannE-book: Phylogenetics in the Genomic Era (Scornavacca et al. 2021) This book was not peer-reviewed by PCI Genomics. It has undergone an internal review by the editors. Accurate reconstructions of the relationships amongst species and the genes encoded in their genomes are an essential foundation for almost all evolutionary inferences emerging from downstream analyses. Molecular phylogenetics has developed as a field over many decades to build suites of models and methods to reconstruct reliable trees that explain, support, or refute such inferences. The genomic era has brought new challenges and opportunities to the field, opening up new areas of research and algorithm development to take advantage of the accumulating large-scale data. Such ‘big-data’ phylogenetics has come to be known as phylogenomics, which broadly aims to connect molecular and evolutionary biology research to address questions centred on relationships amongst taxa, mechanisms of molecular evolution, and the biological functions of genes and other genomic elements. This book brings together experts in the field to present a comprehensive synthesis of Phylogenetics in the Genomic Era, covering key conceptual and methodological aspects of how to build accurate phylogenies and how to apply them in molecular and evolutionary research. The paragraphs below briefly summarise the five constituent parts of the book, highlighting the key concepts, methods, and applications that each part addresses. Being organised in an accessible style, while presenting details to provide depth where necessary, and including guides describing real-world examples of major phylogenomic tools, this collection represents an invaluable resource, particularly for students and newcomers to the field of phylogenomics. Part 1: Phylogenetic analyses in the genomic era Modelling how sequences evolve is a fundamental cornerstone of phylogenetic reconstructions. This part of the book introduces the reader to phylogenetic inference methods and algorithmic optimisations in the contexts of Markov, Maximum Likelihood, and Bayesian models of sequence evolution. The main concepts and theoretical considerations are mapped out for probabilistic Markov models, efficient tree building with Maximum Likelihood methods, and the flexibility and robustness of Bayesian approaches. These are supported with practical examples of phylogenomic applications using the popular tools RAxML and PhyloBayes. By considering theoretical, algorithmic, and practical aspects, these chapters provide readers with a holistic overview of the challenges and recent advances in developing scalable phylogenetic analyses in the genomic era. Part 2: Data quality, model adequacy This part focuses on the importance of considering the appropriateness of the evolutionary models used and the accuracy of the underlying molecular and genomic data. Both these aspects can profoundly affect the results when applying current phylogenomic methods to make inferences about complex biological and evolutionary processes. A clear example is presented for methods for building multiple sequence alignments and subsequent filtering approaches that can greatly impact phylogeny inference. The importance of error detection in (meta)barcode sequencing data is also highlighted, with solutions offered by the MACSE_BARCODE pipeline for accurate taxonomic assignments. Orthology datasets are essential markers for phylogenomic inferences, but the overview of concepts and methods presented shows that they too face challenges with respect to model selection and data quality. Finally, an innovative approach using ancestral gene order reconstructions provides new perspectives on how to assess gene tree accuracy for phylogenomic analyses. By emphasising through examples the importance of using appropriate evolutionary models and assessing input data quality, these chapters alert readers to key limitations that the field as a whole strives to address. Part 3: Resolving phylogenomic conflicts Conflicting phylogenetic signals are commonplace and may derive from statistical or systematic bias. This part of the book addresses possible causes of conflict, discordance between gene trees and species trees and how processes that lead to such conflicts can be described by phylogenetic models. Furthermore, it provides an overview of various models and methods with examples in phylogenomics including their pros and cons. Outlined in detail is the multispecies coalescent model (MSC) and its applications in phylogenomics. An interesting aspect is that different phylogenetic signals leading to conflict are in fact a key source of information rather than a problem that can – and should – be used to point to events like introgression or hybridisation, highlighting possible future trends in this research area. Last but not least, this part of the book also addresses inferring species trees by concatenating single multiple sequence alignments (gene alignments) versus inferring the species tree based on ensembles of single gene trees pointing out advantages and disadvantages of both approaches. As an important take home message from these chapters, it is recommended to be flexible and identify the most appropriate approach for each dataset to be analysed since this may tremendously differ depending on the dataset, setting, taxa, and phylogenetic level addressed by the researcher. Part 4: Functional evolutionary genomics In this part of the book the focus shifts to functional considerations of phylogenomics approaches both in terms of molecular evolution and adaptation and with respect to gene expression. The utility of multi-species analysis is clearly presented in the context of annotating functional genomic elements through quantifying evolutionary constraint and protein-coding potential. An historical perspective on characterising rates of change highlights how phylogenomic datasets help to understand the modes of molecular evolution across the genome, over time, and between lineages. These are contextualised with respect to the specific aim of detecting signatures of adaptation from protein-coding DNA alignments using the example of the MutSelDP-ω∗ model. This is extended with the presentation of the generally rare case of adaptive sequence convergence, where consideration of appropriate models and knowledge of gene functions and phenotypic effects are needed. Constrained or relaxed, selection pressures on sequence or copy-number affect genomic elements in different ways, making the very concept of function difficult to pin down despite it being fundamental to relate the genome to the phenotype and organismal fitness. Here gene expression provides a measurable intermediate, for which the Expression Comparison tool from the Bgee suite allows exploration of expression patterns across multiple animal species taking into account anatomical homology. Overall, phylogenomics applications in functional evolutionary genomics build on a rich theoretical history from molecular analyses where integration with knowledge of gene functions is challenging but critical. Part 5: Phylogenomic applications Rather than attempting to review the full extent of applications linked to phylogenomics, this part of the book focuses on providing detailed specific insights into selected examples and methods concerning i) estimating divergence times, and ii) species delimitation in the era of ‘omics’ data. With respect to estimating divergence times, an exemplary overview is provided for fossil data recovered from geological records, either using fossil data as calibration points with an extant-species-inferred phylogeny, or using a fossilised birth-death process as a mechanistic model that accounts for lineage diversification. Included is a tutorial for a joint approach to infer phylogenies and estimate divergence times using the RevBayes software with various models implemented for different applications and datasets incorporating molecular and morphological data. An interesting excursion is outlined focusing on timescale estimates with respect to viral evolution introducing BEAGLE, a high-performance likelihood-calculation platform that can be used on multi-core systems. As a second major subject, species delimitation is addressed since currently the increasing amount of available genomic data enables extensive inferences, for instance about the degree of genetic isolation among species and ancient and recent introgression events. Describing the history of molecular species delimitation up to the current genomic era and presenting widely used computational methods incorporating single- and multi-locus genomic data, pros and cons are addressed. Finally, a proposal for a new method for delimiting species based on empirical criteria is outlined. In the closing chapter of this part of the book, BPP (Bayesian Markov chain Monte Carlo program) for analysing multi-locus sequence data under the multispecies coalescent (MSC) model with and without introgression is introduced, including a tutorial. These examples together provide accessible details on key conceptual and methodological aspects related to the application of phylogenetics in the genomic era. References Scornavacca C, Delsuc F, Galtier N (2021) Phylogenetics in the Genomic Era. https://hal.inria.fr/PGE/ | Phylogenetics in the Genomic Era | Céline Scornavacca, Frédéric Delsuc, Nicolas Galtier | <p style="text-align: justify;">Molecular phylogenetics was born in the middle of the 20th century, when the advent of protein and DNA sequencing offered a novel way to study the evolutionary relationships between living organisms. The first 50 ye... | | Bacteria and archaea, Bioinformatics, Evolutionary genomics, Functional genomics, Fungi, Plants, Population genomics, Vertebrates, Viruses and transposable elements | Robert Waterhouse | 2022-03-15 17:43:52 | ||
24 Feb 2023
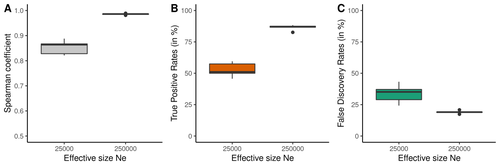
Performance and limitations of linkage-disequilibrium-based methods for inferring the genomic landscape of recombination and detecting hotspots: a simulation studyHow to interpret the inference of recombination landscapes on methods based on linkage disequilibrium?Recommended by Sebastian E. Ramos-Onsins based on reviews by 2 anonymous reviewersData interpretation depends on previously established and validated tools, designed for a specific type of data. These methods, however, are usually based on simple models with validity subject to a set of theoretical parameterized conditions and data types. Accordingly, the tool developers provide the potential users with guidelines for data interpretations within the tools’ limitation. Nevertheless, once the methodology is accepted by the community, it is employed in a large variety of empirical studies outside of the method’s original scope or that typically depart from the standard models used for its design, thus potentially leading to the wrong interpretation of the results. Numerous empirical studies inferred recombination rates across genomes, detecting hotspots of recombination and comparing related species (e.g., Shanfelter et al. 2019, Spence and Song 2019). These studies used indirect methodologies based on the signals that recombination left in the genome, such as linkage disequilibrium and the patterns of haplotype segregation (e.g.,Chan et al. 2012). The conclusions from these analyses have been used, for example, to interpret the evolution of the chromosomal structure or the evolution of recombination among closely related species. Indirect methods have the advantage of collecting a large quantity of recombination events, and thus have a better resolution than direct methods (which only detect the few recombination events occurring at that time). On the other hand, indirect methods are affected by many different evolutionary events, such as demographic changes and selection. Indeed, the inference of recombination levels across the genome has not been studied accurately in non-standard conditions. Linkage disequilibrium is affected by several factors that can modify the recombination inference, such as demographic history, events of selection, population size, and mutation rate, but is also related to the size of the studied sample, and other technical parameters defined for each specific methodology. Raynaud et al (2023) analyzed the reliability of the recombination rate inference when considering the violation of several standard assumptions (evolutionary and methodological) in one of the most popular families of methods based on LDhat (McVean et al. 2004), specifically its improved version, LDhelmet (Chan et al. 2012). These methods cover around 70 % of the studies that infer recombination rates. The authors used recombination maps, obtained from empirical studies on humans, and included hotspots, to perform a detailed simulation study of the capacity of this methodology to correctly infer the pattern of recombination and the location of these hotspots. Correlations between the real, and inferred values from simulations were obtained, as well as several rates, such as the true positive and false discovery rate to detect hotspots. The authors of this work send a message of caution to researchers that are applying this methodology to interpret data from the inference of recombination landscapes and the location of hotspots. The inference of recombination landscapes and hotspots can differ considerably even in standard model conditions. In addition, demographic processes, like bottleneck or admixture, but also the level of population size and mutation rates, can substantially affect the estimation accuracy of the level of recombination and the location of hotspots. Indeed, the inference of the location of hotspots in simulated data with the same landscape, can be very imprecise when standard assumptions are violated or not considered. These effects may lead to incorrect interpretations, for example about the conservation of recombination maps between closely related species. Finally, Raynaud et al (2023) included a useful guide with advice on how to obtain accurate recombination estimations with methods based on linkage disequilibrium, also emphasizing the limitations of such approaches. REFERENCES Chan AH, Jenkins PA, Song YS (2012) Genome-Wide Fine-Scale Recombination Rate Variation in Drosophila melanogaster. PLOS Genetics, 8, e1003090. https://doi.org/10.1371/journal.pgen.1003090 McVean GAT, Myers SR, Hunt S, Deloukas P, Bentley DR, Donnelly P (2004) The Fine-Scale Structure of Recombination Rate Variation in the Human Genome. Science, 304, 581–584. https://doi.org/10.1126/science.1092500 Raynaud M, Gagnaire P-A, Galtier N (2023) Performance and limitations of linkage-disequilibrium-based methods for inferring the genomic landscape of recombination and detecting hotspots: a simulation study. bioRxiv, 2022.03.30.486352, ver. 2 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2022.03.30.486352 Spence JP, Song YS (2019) Inference and analysis of population-specific fine-scale recombination maps across 26 diverse human populations. Science Advances, 5, eaaw9206. https://doi.org/10.1126/sciadv.aaw9206 | Performance and limitations of linkage-disequilibrium-based methods for inferring the genomic landscape of recombination and detecting hotspots: a simulation study | Marie Raynaud, Pierre-Alexandre Gagnaire, Nicolas Galtier | <p style="text-align: justify;">Knowledge of recombination rate variation along the genome provides important insights into genome and phenotypic evolution. Population genomic approaches offer an attractive way to infer the population-scaled recom... | | Bioinformatics, Evolutionary genomics, Population genomics | Sebastian E. Ramos-Onsins | 2022-04-05 14:59:14 |




