
How to interpret the inference of recombination landscapes on methods based on linkage disequilibrium?
 based on reviews by 2 anonymous reviewers
based on reviews by 2 anonymous reviewers
Performance and limitations of linkage-disequilibrium-based methods for inferring the genomic landscape of recombination and detecting hotspots: a simulation study
Abstract
Recommendation: posted 23 February 2023, validated 24 February 2023
Ramos-Onsins, S. (2023) How to interpret the inference of recombination landscapes on methods based on linkage disequilibrium?. Peer Community in Genomics, 100161. https://doi.org/10.24072/pci.genomics.100161
Recommendation
Data interpretation depends on previously established and validated tools, designed for a specific type of data. These methods, however, are usually based on simple models with validity subject to a set of theoretical parameterized conditions and data types. Accordingly, the tool developers provide the potential users with guidelines for data interpretations within the tools’ limitation. Nevertheless, once the methodology is accepted by the community, it is employed in a large variety of empirical studies outside of the method’s original scope or that typically depart from the standard models used for its design, thus potentially leading to the wrong interpretation of the results.
Numerous empirical studies inferred recombination rates across genomes, detecting hotspots of recombination and comparing related species (e.g., Shanfelter et al. 2019, Spence and Song 2019). These studies used indirect methodologies based on the signals that recombination left in the genome, such as linkage disequilibrium and the patterns of haplotype segregation (e.g.,Chan et al. 2012). The conclusions from these analyses have been used, for example, to interpret the evolution of the chromosomal structure or the evolution of recombination among closely related species.
Indirect methods have the advantage of collecting a large quantity of recombination events, and thus have a better resolution than direct methods (which only detect the few recombination events occurring at that time). On the other hand, indirect methods are affected by many different evolutionary events, such as demographic changes and selection. Indeed, the inference of recombination levels across the genome has not been studied accurately in non-standard conditions. Linkage disequilibrium is affected by several factors that can modify the recombination inference, such as demographic history, events of selection, population size, and mutation rate, but is also related to the size of the studied sample, and other technical parameters defined for each specific methodology.
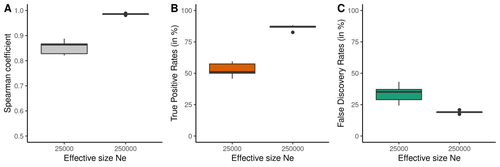
Raynaud et al (2023) analyzed the reliability of the recombination rate inference when considering the violation of several standard assumptions (evolutionary and methodological) in one of the most popular families of methods based on LDhat (McVean et al. 2004), specifically its improved version, LDhelmet (Chan et al. 2012). These methods cover around 70 % of the studies that infer recombination rates. The authors used recombination maps, obtained from empirical studies on humans, and included hotspots, to perform a detailed simulation study of the capacity of this methodology to correctly infer the pattern of recombination and the location of these hotspots. Correlations between the real, and inferred values from simulations were obtained, as well as several rates, such as the true positive and false discovery rate to detect hotspots.
The authors of this work send a message of caution to researchers that are applying this methodology to interpret data from the inference of recombination landscapes and the location of hotspots. The inference of recombination landscapes and hotspots can differ considerably even in standard model conditions. In addition, demographic processes, like bottleneck or admixture, but also the level of population size and mutation rates, can substantially affect the estimation accuracy of the level of recombination and the location of hotspots. Indeed, the inference of the location of hotspots in simulated data with the same landscape, can be very imprecise when standard assumptions are violated or not considered. These effects may lead to incorrect interpretations, for example about the conservation of recombination maps between closely related species. Finally, Raynaud et al (2023) included a useful guide with advice on how to obtain accurate recombination estimations with methods based on linkage disequilibrium, also emphasizing the limitations of such approaches.
REFERENCES
Chan AH, Jenkins PA, Song YS (2012) Genome-Wide Fine-Scale Recombination Rate Variation in Drosophila melanogaster. PLOS Genetics, 8, e1003090. https://doi.org/10.1371/journal.pgen.1003090
McVean GAT, Myers SR, Hunt S, Deloukas P, Bentley DR, Donnelly P (2004) The Fine-Scale Structure of Recombination Rate Variation in the Human Genome. Science, 304, 581–584. https://doi.org/10.1126/science.1092500
Raynaud M, Gagnaire P-A, Galtier N (2023) Performance and limitations of linkage-disequilibrium-based methods for inferring the genomic landscape of recombination and detecting hotspots: a simulation study. bioRxiv, 2022.03.30.486352, ver. 2 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2022.03.30.486352
Spence JP, Song YS (2019) Inference and analysis of population-specific fine-scale recombination maps across 26 diverse human populations. Science Advances, 5, eaaw9206. https://doi.org/10.1126/sciadv.aaw9206

The recommender in charge of the evaluation of the article and the reviewers declared that they have no conflict of interest (as defined in the code of conduct of PCI) with the authors or with the content of the article. The authors declared that they comply with the PCI rule of having no financial conflicts of interest in relation to the content of the article.
This project was funded by the ANR HotRec ANR-19-CE12-0019
Evaluation round #1
DOI or URL of the preprint: https://doi.org/10.1101/2022.03.30.486352
Version of the preprint: 1
Author's Reply, 20 Jan 2023
Decision by Sebastian Ernesto Ramos-Onsins, posted 23 Jun 2022
Dear Nicolas Galtier,
The manuscript titled "Performance and limitations of linkage-disequilibrium-based methods for inferring the genomic landscape of recombination and detecting hotspots: a simulation study" have been reviewed.
The two reviewers find this manuscript interesting and well written. The results obtained from the simulation study provide valuable information for the interpretation and understanding of the empirical data. The reviewers have provided various comments and suggestions that will help improve the manuscript. Specifically, they are both interested in understanding the impact of additional evolutionary parameters, such as the effect of demography and positive and negative selection.
Other minor corrections should be considered. Some figures are color coded although the color is not visible on the graph (Figure 3, Figure S3), Other Figures have unclear comparisons (Figure 5, the actual rate is hardly visible in blue) and some others may include labels additions for a quicker understanding of the multiple axis. Improve the presentation of the figure in its revised version.
I found some typos or unclear sentences (line 274, should the sensitivity be TPR?, line 275, FDR is a way to measure type I error, which is based on alternative hypotheses, although type I error is usually defined as FPR).
Consider and respond to all comments and suggestions from reviewers before submitting the new version.
Sincerely,
Sebastian E. Ramos-Onsins
Reviewed by anonymous reviewer 1, 25 May 2022
In this article, Raynaud et al. evaluate the ability of a population genomic and linkage-disequilibrium based approach LDhelmet for inferring biologically-realistic landscapes of recombination in humans. In particular, using extensive simulations, they evaluate the accuracy of detecting recombination hotspots and discuss implications for prior studies that have used such approaches to address questions such as whether recombination hotspots are evolutionarily conserved between closely related species.
They conclude that while LD-based approaches can provide high quality recombination maps in species with simple recombination landscapes, for species with complex recombinational landscapes their usage for biological interpretations regarding the evolutionary conservation of recombination maps warrants more caution. The biases and uncertainty in inferred recombination rates as well as the potential for false positives and false negatives in hotspot detection needs to be taken into account before making these interpretations. Furthermore, they note that simulation scenarios in this paper are optimistic in terms of the reliabiity of hotspot detection and in empirical data there are further sources of noise including sequencing, mapping and phasing errors as well population demography.
Comments:
1. Based on empirical data and error rates, would it be possible to include some plausible scenarios of sequencing error? For accounting for uncertainty of phasing, can genotype data be simulated and subsequenly phased before conducting analysis.
2. There could additional confounding factors when inferring fine-scale recombination patterns from LD, in particular the confounding between cross-overs and gene-conversions. Has any analyses been done in this regard?
3. Human populations can have complex demography with migration between populations. Can this further impact the accuracy of such inference methods or do we expect relative rates to be robust?
4. Presence of natural selection in a region can bias recombination inference. How may this affect hotspot inference? In general, other than simple recombination landscapes, what other assumptions need to be met for hotspot inference to be accurate?
5. Structural variation such as inversions can also impact recombination inference in certain species like drosophilia. This may further contribute to uncertainty.
https://doi.org/10.24072/pci.genomics.100161.rev11Reviewed by anonymous reviewer 2, 08 Jun 2022
Raynaud et al present a manuscript using simulations to test the performance and limitations of a commonly used method to infer recombination landscapes, LDhelmet. They find that maps produced with the method have good correlations with the true simulated maps, but there are limitations when the method is applied to detect hotspots. In particular, LDhelmet tends to overestimate the local recombination rate. Additionally, they note that the method can find shared hotspots when there are no real shared hotspots. This result has implications, for example, for interpreting data from Shanfelter et al 2019, who used LDhelmet and found little overlap between marine and freshwater populations of three-spine stickleback. In general I found the manuscript interesting and well-written, but I have some suggestions which I hope will improve the manuscript.
Major comments:
· I found the scope of the study to be more limited that I expected. The authors focus on a single method published in 2012. While this method is widely used, there have been several additional methods published in the following years (which the authors cite in the introduction, line 62-63). I would be interested in understanding how other recently developed methods compare.
· Additional evolutionary parameters: in a study like this, one has to make choices about which parameters to study. I agree with the authors that studying the impacts of effective population size, mutation rates, and recombination rates is important. However, I will suggest 2 additional factors that I think would be substantial benefits to the manuscript:
a. Selection: the impacts of both positive and negative selection on patterns of LD are well known. However, I wonder how these forces affect hotspot inference. The authors could implement simple simulations in SLiM (Haller et al 2019 MBE), which will output a VCF and should fit fairly smoothly into the pipeline the authors have already set up.
b. Demographic changes with large effects on LD: it is well known that bottlenecks and exponential growth will affect LD patterns. Given the results presented in the paper, I would expect that these would also affect inferences of recombination hotspots, but I would be interested in quantifying how much.
Minor comments:
· Could the authors give some intuition about what the block penalty does?
· Line 338-339: 10-9 should be 10^-9




