
Latest recommendations

| Id | Title * | Authors * | Abstract * | Picture * | Thematic fields * ▲ | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
08 Apr 2022

POSTPRINT
Phylogenetics in the Genomic Era“Phylogenetics in the Genomic Era” brings together experts in the field to present a comprehensive synthesisRecommended by Robert Waterhouse and Karen MeusemannE-book: Phylogenetics in the Genomic Era (Scornavacca et al. 2021) This book was not peer-reviewed by PCI Genomics. It has undergone an internal review by the editors. Accurate reconstructions of the relationships amongst species and the genes encoded in their genomes are an essential foundation for almost all evolutionary inferences emerging from downstream analyses. Molecular phylogenetics has developed as a field over many decades to build suites of models and methods to reconstruct reliable trees that explain, support, or refute such inferences. The genomic era has brought new challenges and opportunities to the field, opening up new areas of research and algorithm development to take advantage of the accumulating large-scale data. Such ‘big-data’ phylogenetics has come to be known as phylogenomics, which broadly aims to connect molecular and evolutionary biology research to address questions centred on relationships amongst taxa, mechanisms of molecular evolution, and the biological functions of genes and other genomic elements. This book brings together experts in the field to present a comprehensive synthesis of Phylogenetics in the Genomic Era, covering key conceptual and methodological aspects of how to build accurate phylogenies and how to apply them in molecular and evolutionary research. The paragraphs below briefly summarise the five constituent parts of the book, highlighting the key concepts, methods, and applications that each part addresses. Being organised in an accessible style, while presenting details to provide depth where necessary, and including guides describing real-world examples of major phylogenomic tools, this collection represents an invaluable resource, particularly for students and newcomers to the field of phylogenomics. Part 1: Phylogenetic analyses in the genomic era Modelling how sequences evolve is a fundamental cornerstone of phylogenetic reconstructions. This part of the book introduces the reader to phylogenetic inference methods and algorithmic optimisations in the contexts of Markov, Maximum Likelihood, and Bayesian models of sequence evolution. The main concepts and theoretical considerations are mapped out for probabilistic Markov models, efficient tree building with Maximum Likelihood methods, and the flexibility and robustness of Bayesian approaches. These are supported with practical examples of phylogenomic applications using the popular tools RAxML and PhyloBayes. By considering theoretical, algorithmic, and practical aspects, these chapters provide readers with a holistic overview of the challenges and recent advances in developing scalable phylogenetic analyses in the genomic era. Part 2: Data quality, model adequacy This part focuses on the importance of considering the appropriateness of the evolutionary models used and the accuracy of the underlying molecular and genomic data. Both these aspects can profoundly affect the results when applying current phylogenomic methods to make inferences about complex biological and evolutionary processes. A clear example is presented for methods for building multiple sequence alignments and subsequent filtering approaches that can greatly impact phylogeny inference. The importance of error detection in (meta)barcode sequencing data is also highlighted, with solutions offered by the MACSE_BARCODE pipeline for accurate taxonomic assignments. Orthology datasets are essential markers for phylogenomic inferences, but the overview of concepts and methods presented shows that they too face challenges with respect to model selection and data quality. Finally, an innovative approach using ancestral gene order reconstructions provides new perspectives on how to assess gene tree accuracy for phylogenomic analyses. By emphasising through examples the importance of using appropriate evolutionary models and assessing input data quality, these chapters alert readers to key limitations that the field as a whole strives to address. Part 3: Resolving phylogenomic conflicts Conflicting phylogenetic signals are commonplace and may derive from statistical or systematic bias. This part of the book addresses possible causes of conflict, discordance between gene trees and species trees and how processes that lead to such conflicts can be described by phylogenetic models. Furthermore, it provides an overview of various models and methods with examples in phylogenomics including their pros and cons. Outlined in detail is the multispecies coalescent model (MSC) and its applications in phylogenomics. An interesting aspect is that different phylogenetic signals leading to conflict are in fact a key source of information rather than a problem that can – and should – be used to point to events like introgression or hybridisation, highlighting possible future trends in this research area. Last but not least, this part of the book also addresses inferring species trees by concatenating single multiple sequence alignments (gene alignments) versus inferring the species tree based on ensembles of single gene trees pointing out advantages and disadvantages of both approaches. As an important take home message from these chapters, it is recommended to be flexible and identify the most appropriate approach for each dataset to be analysed since this may tremendously differ depending on the dataset, setting, taxa, and phylogenetic level addressed by the researcher. Part 4: Functional evolutionary genomics In this part of the book the focus shifts to functional considerations of phylogenomics approaches both in terms of molecular evolution and adaptation and with respect to gene expression. The utility of multi-species analysis is clearly presented in the context of annotating functional genomic elements through quantifying evolutionary constraint and protein-coding potential. An historical perspective on characterising rates of change highlights how phylogenomic datasets help to understand the modes of molecular evolution across the genome, over time, and between lineages. These are contextualised with respect to the specific aim of detecting signatures of adaptation from protein-coding DNA alignments using the example of the MutSelDP-ω∗ model. This is extended with the presentation of the generally rare case of adaptive sequence convergence, where consideration of appropriate models and knowledge of gene functions and phenotypic effects are needed. Constrained or relaxed, selection pressures on sequence or copy-number affect genomic elements in different ways, making the very concept of function difficult to pin down despite it being fundamental to relate the genome to the phenotype and organismal fitness. Here gene expression provides a measurable intermediate, for which the Expression Comparison tool from the Bgee suite allows exploration of expression patterns across multiple animal species taking into account anatomical homology. Overall, phylogenomics applications in functional evolutionary genomics build on a rich theoretical history from molecular analyses where integration with knowledge of gene functions is challenging but critical. Part 5: Phylogenomic applications Rather than attempting to review the full extent of applications linked to phylogenomics, this part of the book focuses on providing detailed specific insights into selected examples and methods concerning i) estimating divergence times, and ii) species delimitation in the era of ‘omics’ data. With respect to estimating divergence times, an exemplary overview is provided for fossil data recovered from geological records, either using fossil data as calibration points with an extant-species-inferred phylogeny, or using a fossilised birth-death process as a mechanistic model that accounts for lineage diversification. Included is a tutorial for a joint approach to infer phylogenies and estimate divergence times using the RevBayes software with various models implemented for different applications and datasets incorporating molecular and morphological data. An interesting excursion is outlined focusing on timescale estimates with respect to viral evolution introducing BEAGLE, a high-performance likelihood-calculation platform that can be used on multi-core systems. As a second major subject, species delimitation is addressed since currently the increasing amount of available genomic data enables extensive inferences, for instance about the degree of genetic isolation among species and ancient and recent introgression events. Describing the history of molecular species delimitation up to the current genomic era and presenting widely used computational methods incorporating single- and multi-locus genomic data, pros and cons are addressed. Finally, a proposal for a new method for delimiting species based on empirical criteria is outlined. In the closing chapter of this part of the book, BPP (Bayesian Markov chain Monte Carlo program) for analysing multi-locus sequence data under the multispecies coalescent (MSC) model with and without introgression is introduced, including a tutorial. These examples together provide accessible details on key conceptual and methodological aspects related to the application of phylogenetics in the genomic era. References Scornavacca C, Delsuc F, Galtier N (2021) Phylogenetics in the Genomic Era. https://hal.inria.fr/PGE/ | Phylogenetics in the Genomic Era | Céline Scornavacca, Frédéric Delsuc, Nicolas Galtier | <p style="text-align: justify;">Molecular phylogenetics was born in the middle of the 20th century, when the advent of protein and DNA sequencing offered a novel way to study the evolutionary relationships between living organisms. The first 50 ye... | | Bacteria and archaea, Bioinformatics, Evolutionary genomics, Functional genomics, Fungi, Plants, Population genomics, Vertebrates, Viruses and transposable elements | Robert Waterhouse | 2022-03-15 17:43:52 | ||
07 Feb 2023
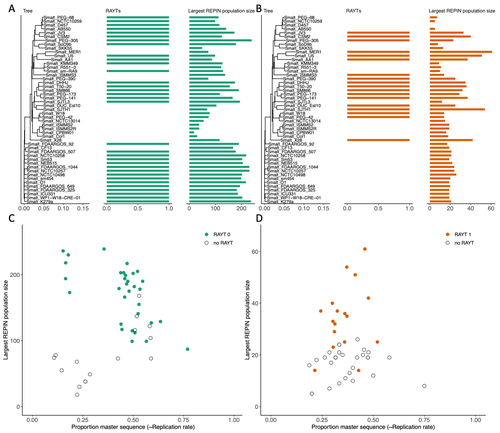
RAREFAN: A webservice to identify REPINs and RAYTs in bacterial genomesA workflow for studying enigmatic non-autonomous transposable elements across bacteriaRecommended by Gavin Douglas based on reviews by Sophie Abby and 1 anonymous reviewer based on reviews by Sophie Abby and 1 anonymous reviewer
Repetitive extragenic palindromic sequences (REPs) are common repetitive elements in bacterial genomes (Gilson et al., 1984; Stern et al., 1984). In 2011, Bertels and Rainey identified that REPs are overrepresented in pairs of inverted repeats, which likely form hairpin structures, that they referred to as “REP doublets forming hairpins” (REPINs). Based on bioinformatics analyses, they argued that REPINs are likely selfish elements that evolved from REPs flanking particular transposes (Bertels and Rainey, 2011). These transposases, so-called REP-associated tyrosine transposases (RAYTs), were known to be highly associated with the REP content in a genome and to have characteristic upstream and downstream flanking REPs (Nunvar et al., 2010). The flanking REPs likely enable RAYT transposition, and their horizontal replication is physically linked to this process. In contrast, Bertels and Rainey hypothesized that REPINs are selfish elements that are highly replicated due to the similarity in arrangement to these RAYT-flanking REPs, but independent of RAYT transposition and generally with no impact on bacterial fitness (Bertels and Rainey, 2011). This last point was especially contentious, as REPINs are highly conserved within species (Bertels and Rainey, 2023), which is unusual for non-beneficial bacterial DNA (Mira et al., 2001). Bertels and Rainey have since refined their argument to be that REPINs must provide benefits to host cells, but that there are nonetheless signatures of intragenomic conflict in genomes associated with these elements (Bertels and Rainey, 2023). These signatures reflect the divergent levels of selections driving REPIN distribution: selection at the level of each DNA element and selection on each individual bacterium. I found this observation particularly interesting as I and my colleague recently argued that these divergent levels of selection, and the interaction between them, is key to understanding bacterial pangenome diversity (Douglas and Shapiro, 2021). REPINs could be an excellent system for investigating these levels of selection across bacteria more generally. The problem is that REPINs have not been widely characterized in bacterial genomes, partially because no bioinformatic workflow has been available for this purpose. To address this problem, Fortmann-Grote et al. (2023) developed RAREFAN, which is a web server for identifying RAYTs and associated REPINs in a set of input genomes. The authors showcase their tool by applying it to 49 Stenotrophomonas maltophilia genomes and providing examples of how to identify and assess RAYT-REPIN hits. The workflow requires several manual steps, but nonetheless represents a straightforward and standardized approach. Overall, this workflow should enable RAYTs and REPINs to be identified across diverse bacterial species, which will facilitate further investigation into the mechanisms driving their maintenance and spread. References Bertels F, Rainey PB (2023) Ancient Darwinian replicators nested within eubacterial genomes. BioEssays, 45, 2200085. https://doi.org/10.1002/bies.202200085 Bertels F, Rainey PB (2011) Within-Genome Evolution of REPINs: a New Family of Miniature Mobile DNA in Bacteria. PLOS Genetics, 7, e1002132. https://doi.org/10.1371/journal.pgen.1002132 Douglas GM, Shapiro BJ (2021) Genic Selection Within Prokaryotic Pangenomes. Genome Biology and Evolution, 13, evab234. https://doi.org/10.1093/gbe/evab234 Fortmann-Grote C, Irmer J von, Bertels F (2023) RAREFAN: A webservice to identify REPINs and RAYTs in bacterial genomes. bioRxiv, 2022.05.22.493013, ver. 4 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2022.05.22.493013 Gilson E, Clément J m., Brutlag D, Hofnung M (1984) A family of dispersed repetitive extragenic palindromic DNA sequences in E. coli. The EMBO Journal, 3, 1417–1421. https://doi.org/10.1002/j.1460-2075.1984.tb01986.x Mira A, Ochman H, Moran NA (2001) Deletional bias and the evolution of bacterial genomes. Trends in Genetics, 17, 589–596. https://doi.org/10.1016/S0168-9525(01)02447-7 Nunvar J, Huckova T, Licha I (2010) Identification and characterization of repetitive extragenic palindromes (REP)-associated tyrosine transposases: implications for REP evolution and dynamics in bacterial genomes. BMC Genomics, 11, 44. https://doi.org/10.1186/1471-2164-11-44 Stern MJ, Ames GF-L, Smith NH, Clare Robinson E, Higgins CF (1984) Repetitive extragenic palindromic sequences: A major component of the bacterial genome. Cell, 37, 1015–1026. https://doi.org/10.1016/0092-8674(84)90436-7 | RAREFAN: A webservice to identify REPINs and RAYTs in bacterial genomes | Frederic Bertels, Julia von Irmer, Carsten Fortmann-Grote | <p style="text-align: justify;">Compared to eukaryotes, repetitive sequences are rare in bacterial genomes and usually do not persist for long. Yet, there is at least one class of persistent prokaryotic mobile genetic elements: REPINs. REPINs are ... | | Bacteria and archaea, Bioinformatics, Evolutionary genomics, Viruses and transposable elements | Gavin Douglas | 2022-06-07 08:21:34 | ||
24 Feb 2023

MacSyFinder v2: Improved modelling and search engine to identify molecular systems in genomesA unique and customizable approach for functionally annotating prokaryotic genomesRecommended by Gavin Douglas based on reviews by Kwee Boon Brandon Seah and Max Emil Schön
Macromolecular System Finder (MacSyFinder) v2 (Néron et al., 2023) is a newly updated approach for performing functional annotation of prokaryotic genomes (Abby et al., 2014). This tool parses an input file of protein sequences from a single genome (either ordered by genome location or unordered) and identifies the presence of specific cellular functions (referred to as “systems”). These systems are called based on two criteria: (1) that the "quorum" of a minimum set of core proteins involved is reached the “quorum” of a minimum set of core proteins being involved that are present, and (2) that the genes encoding these proteins are in the expected genomic organization (e.g., within the same order in an operon), when ordered data is provided. I believe the MacSyFinder approach represents an improvement over more commonly used methods exactly because it can incorporate such information on genomic organization, and also because it is more customizable. Before properly appreciating these points, it is worth noting the norms and key challenges surrounding high-throughput functional annotation of prokaryotic genomes. Genome sequences are being added to online repositories at increasing rates, which has led to an enormous amount of bacterial genome diversity available to investigate (Altermann et al., 2022). A key aspect of understanding this diversity is the functional annotation step, which enables genes to be grouped into more biologically interpretable categories. For instance, gene calls can be mapped against existing Clusters of Orthologous Genes, which are themselves grouped into general categories such as ‘Transcription’ and ‘Lipid metabolism’ (Galperin et al., 2021). This approach is valuable but is primarily used for global summaries of functional annotations within a genome: for example, it could be useful to know that a genome is particularly enriched for genes involved in lipid metabolism. However, knowing that a particular gene is involved in the general process of lipid metabolism is less likely to be actionable. In other words, the desired specificity of a gene’s functional annotation will depend on the exact question being investigated. There is no shortage of functional ontologies in genomics that can be applied for this purpose (Douglas and Langille, 2021), and researchers are often overwhelmed by the choice of which functional ontology to use. In this context, giving researchers the ability to precisely specify the gene families and operon structures they are interested in identifying across genomes provides useful control over what precise functions they are profiling. Of course, most researchers will lack the information and/or expertise to fully take advantage of MacSyFinder’s customizable features, but having this option for specialized purposes is valuable. The other MacSyFinder feature that I find especially noteworthy is that it can incorporate genomic organization (e.g., of genes ordered in operons) when calling systems. This is a rare feature among commonly used tools for functional annotation and likely results in much higher specificity. As the authors note, this capability makes the co-occurrence of paralogs, and other divergent genes that share sequence similarity, to contribute less noise (i.e., they result in fewer false positive calls). It is important to emphasize that these features are not new additions in MacSyFinder v2, but there are many other valuable changes. Most practically, this release is written in Python 3, rather than the obsolete Python 2.7, and was made more computationally efficient, which will enable MacSyFinder to be more widely used and more easily maintained moving forward. In addition, the search algorithm for analyzing individual proteins was fundamentally updated as well. The authors show that their improvements to the search algorithm result in an 8% and 20% increase in the number of identified calls for single and multi-locus secretion systems, respectively. Taken together, MacSyFinder v2 represents both practical and scientific improvements over the previous version, which will be of great value to the field. References Abby SS, Néron B, Ménager H, Touchon M, Rocha EPC (2014) MacSyFinder: A Program to Mine Genomes for Molecular Systems with an Application to CRISPR-Cas Systems. PLOS ONE, 9, e110726. https://doi.org/10.1371/journal.pone.0110726 Altermann E, Tegetmeyer HE, Chanyi RM (2022) The evolution of bacterial genome assemblies - where do we need to go next? Microbiome Research Reports, 1, 15. https://doi.org/10.20517/mrr.2022.02 Douglas GM, Langille MGI (2021) A primer and discussion on DNA-based microbiome data and related bioinformatics analyses. Peer Community Journal, 1. https://doi.org/10.24072/pcjournal.2 Galperin MY, Wolf YI, Makarova KS, Vera Alvarez R, Landsman D, Koonin EV (2021) COG database update: focus on microbial diversity, model organisms, and widespread pathogens. Nucleic Acids Research, 49, D274–D281. https://doi.org/10.1093/nar/gkaa1018 Néron B, Denise R, Coluzzi C, Touchon M, Rocha EPC, Abby SS (2023) MacSyFinder v2: Improved modelling and search engine to identify molecular systems in genomes. bioRxiv, 2022.09.02.506364, ver. 2 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2022.09.02.506364 | MacSyFinder v2: Improved modelling and search engine to identify molecular systems in genomes | Bertrand Néron, Rémi Denise, Charles Coluzzi, Marie Touchon, Eduardo P. C. Rocha, Sophie S. Abby | <p style="text-align: justify;">Complex cellular functions are usually encoded by a set of genes in one or a few organized genetic loci in microbial genomes. Macromolecular System Finder (MacSyFinder) is a program that uses these properties to mod... | | Bacteria and archaea, Bioinformatics, Functional genomics | Gavin Douglas | Kwee Boon Brandon Seah, Max Emil Schön | 2022-09-09 10:30:31 | |
16 Dec 2022
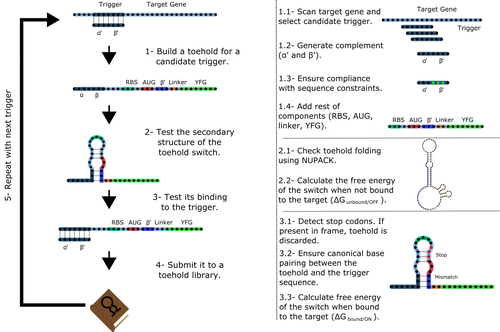
Toeholder: a Software for Automated Design and In Silico Validation of Toehold RiboswitchesA novel approach for engineering biological systems by interfacing computer science with synthetic biologyRecommended by Sahar Melamed based on reviews by Wim Wranken and 1 anonymous reviewerBiological systems depend on finely tuned interactions of their components. Thus, regulating these components is critical for the system's functionality. In prokaryotic cells, riboswitches are regulatory elements controlling transcription or translation. Riboswitches are RNA molecules that are usually located in the 5′-untranslated region of protein-coding genes. They generate secondary structures leading to the regulation of the expression of the downstream protein-coding gene (Kavita and Breaker, 2022). Riboswitches are very versatile and can bind a wide range of small molecules; in many cases, these are metabolic byproducts from the gene’s enzymatic or signaling pathway. Their versatility and abundance in many species make them attractive for synthetic biological circuits. One class that has been drawing the attention of synthetic biologists is toehold switches (Ekdahl et al., 2022; Green et al., 2014). These are single-stranded RNA molecules harboring the necessary elements for translation initiation of the downstream gene: a ribosome-binding site and a start codon. Conformation change of toehold switches is triggered by an RNA molecule, which enables translation. To exploit the most out of toehold switches, automation of their design would be highly advantageous. Cisneros and colleagues (Cisneros et al., 2022) developed a tool, “Toeholder”, that automates the design of toehold switches and performs in silico tests to select switch candidates for a target gene. Toeholder is an open-source tool that provides a comprehensive and automated workflow for the design of toehold switches. While web tools have been developed for designing toehold switches (To et al., 2018), Toeholder represents an intriguing approach to engineering biological systems by coupling synthetic biology with computational biology. Using molecular dynamics simulations, it identified the positions in the toehold switch where hydrogen bonds fluctuate the most. Identifying these regions holds great potential for modifications when refining the design of the riboswitches. To be effective, toehold switches should provide a strong ON signal and a weak OFF signal in the presence or the absence of a target, respectively. Toeholder nicely ranks the candidate toehold switches based on experimental evidence that correlates with toehold performance (based on good ON/OFF ratios). Riboswitches are highly appealing for a broad range of applications, including pharmaceutical and medical purposes (Blount and Breaker, 2006; Giarimoglou et al., 2022; Tickner and Farzan, 2021), thanks to their adaptability and inexpensiveness. The Toeholder tool developed by Cisneros and colleagues is expected to promote the implementation of toehold switches into these various applications. References Blount KF, Breaker RR (2006) Riboswitches as antibacterial drug targets. Nature Biotechnology, 24, 1558–1564. https://doi.org/10.1038/nbt1268 Cisneros AF, Rouleau FD, Bautista C, Lemieux P, Dumont-Leblond N, ULaval 2019 T iGEM (2022) Toeholder: a Software for Automated Design and In Silico Validation of Toehold Riboswitches. bioRxiv, 2021.11.09.467922, ver. 3 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2021.11.09.467922 Ekdahl AM, Rojano-Nisimura AM, Contreras LM (2022) Engineering Toehold-Mediated Switches for Native RNA Detection and Regulation in Bacteria. Journal of Molecular Biology, 434, 167689. https://doi.org/10.1016/j.jmb.2022.167689 Giarimoglou N, Kouvela A, Maniatis A, Papakyriakou A, Zhang J, Stamatopoulou V, Stathopoulos C (2022) A Riboswitch-Driven Era of New Antibacterials. Antibiotics, 11, 1243. https://doi.org/10.3390/antibiotics11091243 Green AA, Silver PA, Collins JJ, Yin P (2014) Toehold Switches: De-Novo-Designed Regulators of Gene Expression. Cell, 159, 925–939. https://doi.org/10.1016/j.cell.2014.10.002 Kavita K, Breaker RR (2022) Discovering riboswitches: the past and the future. Trends in Biochemical Sciences. https://doi.org/10.1016/j.tibs.2022.08.009 Tickner ZJ, Farzan M (2021) Riboswitches for Controlled Expression of Therapeutic Transgenes Delivered by Adeno-Associated Viral Vectors. Pharmaceuticals, 14, 554. https://doi.org/10.3390/ph14060554 To AC-Y, Chu DH-T, Wang AR, Li FC-Y, Chiu AW-O, Gao DY, Choi CHJ, Kong S-K, Chan T-F, Chan K-M, Yip KY (2018) A comprehensive web tool for toehold switch design. Bioinformatics, 34, 2862–2864. https://doi.org/10.1093/bioinformatics/bty216 | Toeholder: a Software for Automated Design and In Silico Validation of Toehold Riboswitches | Angel F. Cisneros, François D. Rouleau, Carla Bautista, Pascale Lemieux, Nathan Dumont-Leblond | <p>Abstract: Synthetic biology aims to engineer biological circuits, which often involve gene expression. A particularly promising group of regulatory elements are riboswitches because of their versatility with respect to their targets, but e... | | Bioinformatics | Sahar Melamed | 2022-02-16 14:40:13 | ||
18 Jul 2022
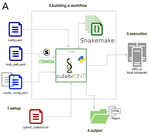
CulebrONT: a streamlined long reads multi-assembler pipeline for prokaryotic and eukaryotic genomesA flexible and reproducible pipeline for long-read assembly and evaluationRecommended by Raúl Castanera based on reviews by Benjamin Istace and Valentine MurigneuxThird-generation sequencing has revolutionised de novo genome assembly. Thanks to this technology, genome reference sequences have evolved from fragmented drafts to gapless, telomere-to-telomere genome assemblies. Long reads produced by Oxford Nanopore and PacBio technologies can span structural variants and resolve complex repetitive regions such as centromeres, unlocking previously inaccessible genomic information. Nowadays, many research groups can afford to sequence the genome of their working model using long reads. Nevertheless, genome assembly poses a significant computational challenge. Read length, quality, coverage and genomic features such as repeat content can affect assembly contiguity, accuracy, and completeness in almost unpredictable ways. Consequently, there is no best universal software or protocol for this task. Producing a high-quality assembly requires chaining several tools into pipelines and performing extensive comparisons between the assemblies obtained by different tool combinations to decide which one is the best. This task can be extremely challenging, as the number of tools available rises very rapidly, and thorough benchmarks cannot be updated and published at such a fast pace. In their paper, Orjuela and collaborators present CulebrONT [1], a universal pipeline that greatly contributes to overcoming these challenges and facilitates long-read genome assembly for all taxonomic groups. CulebrONT incorporates six commonly used assemblers and allows to perform assembly, circularization (if needed), polishing, and evaluation in a simple framework. One important aspect of CulebrONT is its modularity, which allows the activation or deactivation of specific tools, giving great flexibility to the user. Nevertheless, possibly the best feature of CulebrONT is the opportunity to benchmark the selected tool combinations based on the excellent report generated by the pipeline. This HTML report aggregates the output of several tools for quality evaluation of the assemblies (e.g. BUSCO [2] or QUAST [3]) generated by the different assemblers, in addition to the running time and configuration parameters. Such information is of great help to identify the best-suited pipeline, as exemplified by the authors using four datasets of different taxonomic origins. Finally, CulebrONT can handle multiple samples in parallel, which makes it a good solution for laboratories looking for multiple assemblies on a large scale. References 1. Orjuela J, Comte A, Ravel S, Charriat F, Vi T, Sabot F, Cunnac S (2022) CulebrONT: a streamlined long reads multi-assembler pipeline for prokaryotic and eukaryotic genomes. bioRxiv, 2021.07.19.452922, ver. 5 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2021.07.19.452922 2. Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM (2015) BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics, 31, 3210–3212. https://doi.org/10.1093/bioinformatics/btv351 3. Gurevich A, Saveliev V, Vyahhi N, Tesler G (2013) QUAST: quality assessment tool for genome assemblies. Bioinformatics, 29, 1072–1075. https://doi.org/10.1093/bioinformatics/btt086 | CulebrONT: a streamlined long reads multi-assembler pipeline for prokaryotic and eukaryotic genomes | Julie Orjuela, Aurore Comte, Sébastien Ravel, Florian Charriat, Tram Vi, Francois Sabot, Sébastien Cunnac | <p style="text-align: justify;">Using long reads provides higher contiguity and better genome assemblies. However, producing such high quality sequences from raw reads requires to chain a growing set of tools, and determining the best workflow is ... | | Bioinformatics | Raúl Castanera | Valentine Murigneux | 2022-02-22 16:21:25 | |
14 Sep 2023
Expression of cell-wall related genes is highly variable and correlates with sepal morphologyThe same but different: How small scale hidden variations can have large effectsRecommended by Francois Sabot based on reviews by Sandra Corjito and 1 anonymous reviewer
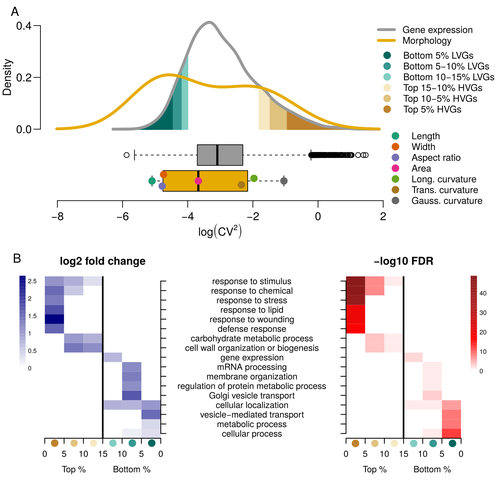
For ages, we considered only single genes, or just a few, in order to understand the relationship between phenotype and genotype in response to environmental challenges. Recently, the use of meaningful groups of genes, e.g. gene regulatory networks, or modules of co-expression, allowed scientists to have a larger view of gene regulation. However, all these findings were based on contrasted genotypes, e.g. between wild-types and mutants, as the implicit assumption often made is that there is little transcriptomic variability within the same genotype context. Hartasànchez and collaborators (2023) decided to challenge both views: they used a single genotype instead of two, the famous A. thaliana Col0, and numerous plants, and considered whole gene networks related to sepal morphology and its variations. They used a clever approach, combining high-level phenotyping and gene expression to better understand phenomena and regulations underlying sepal morphologies. Using multiple controls, they showed that basic variations in the expression of genes related to the cell wall regulation, as well as the ones involved in chloroplast metabolism, influenced the global transcriptomic pattern observed in sepal while being in near-identical genetic background and controlling for all other experimental conditions. The paper of Hartasànchez et al. is thus a tremendous call for humility in biology, as we saw in their work that we just understand the gross machinery. However, the Devil is in the details: understanding those very small variations that may have a large influence on phenotypes, and thus on local adaptation to environmental challenges, is of great importance in these times of climatic changes. References Hartasánchez DA, Kiss A, Battu V, Soraru C, Delgado-Vaquera A, Massinon F, Brasó-Vives M, Mollier C, Martin-Magniette M-L, Boudaoud A, Monéger F. 2023. Expression of cell-wall related genes is highly variable and correlates with sepal morphology. bioRxiv, ver. 4, peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2022.04.26.489498 | Expression of cell-wall related genes is highly variable and correlates with sepal morphology | Diego A. Hartasánchez, Annamaria Kiss, Virginie Battu, Charline Soraru, Abigail Delgado-Vaquera, Florian Massinon, Marina Brasó-Vives, Corentin Mollier, Marie-Laure Martin-Magniette, Arezki Boudaoud, Françoise Monéger | <p style="text-align: justify;">Control of organ morphology is a fundamental feature of living organisms. There is, however, observable variation in organ size and shape within a given genotype. Taking the sepal of Arabidopsis as a model, we inves... | | Bioinformatics, Epigenomics, Plants | Francois Sabot | 2023-03-14 19:10:15 | ||
11 May 2024
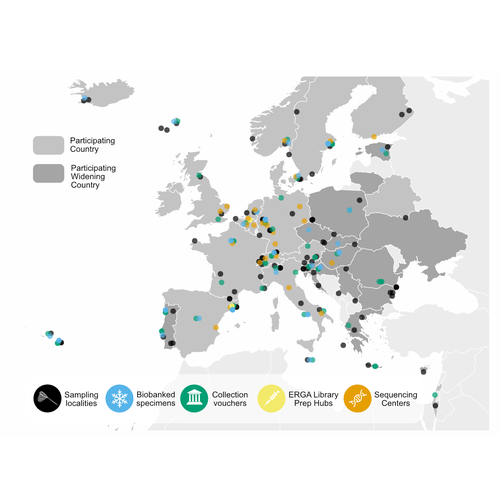
The European Reference Genome Atlas: piloting a decentralised approach to equitable biodiversity genomicsInformed Choices, Cohesive Future: Decisions and Recommendations for ERGARecommended by Jitendra Narayan based on reviews by Justin Ideozu and Eric Crandall
The European Reference Genome Atlas (ERGA) (Mc Cartney et al, 2024, Mazzoni et al, 2023) demonstrates the collaborative spirit and intellectual abilities of researchers from 33 European countries. This ambitious project, which is part of the Earth BioGenome Project (Lewin et al., 2018) Phase II, has embarked on an unprecedented mission: to decipher the genetic makeup of 150,000 species over a span of four years. At the heart of ERGA is a decentralized pilot infrastructure specifically built to assist the production of high-quality reference genomes. This structure acts as a scaffold for the massive task of genome sequencing, giving the necessary framework to manage the complexity of genomic research. The research paper under consideration offers a comprehensive narrative of ERGA's evolution, outlining both successes and challenges encountered along the road. One of the most significant issues addressed in the manuscript is the equitable distribution of resources and expertise among participating laboratories and countries. In a project of this magnitude, it is critical to leverage the pooled talents and capacities of researchers from across Europe. ERGA's pan-European network promotes communications and collaboration, creating an environment in which knowledge flows freely and barriers are overcome. This adoption of strong coordination and communication tactics will be essential to ERGA's success. Scientific collaboration depends on efficient communication channels because they allow researchers to share resources, collaborate on new initiatives, and exchange ideas. Through a diverse range of gatherings, courses, and virtual discussion boards, ERGA fosters an environment of transparency and cooperation among members, enabling scientists to overcome challenges and make significant discoveries. The importance ERGA places on training and information transfer programmes is a pillar of its strategy. Understanding the importance of capacity development, ERGA invests in providing researchers with the knowledge and abilities necessary for effectively navigating the complicated terrain of genomic research. A wide range of subjects are covered in training programmes (Larivière et al. 2023), from sample preparation and collection to data processing methods and sequencing technology. Through the development of a group of highly qualified experts, ERGA creates the foundation for continued advancement and creativity in the genomics sector. This manuscript also covers in detail the technological workflows and sequencing techniques used in ERGA's pilot infrastructure. With the aid of cutting-edge sequencing technologies based on both long-read and short-read sequencing, they are working to unravel the complex structure of the genetic code with a level of accuracy and precision never before possible. To guarantee the accuracy of genetic data and prevent mistakes and flaws that can jeopardize the findings' integrity, quality control methods are put in place. Despite having a focus on genome sequencing due to its technological complexities, ERGA also remains firm in its dedication to metadata collection and sample validation. Metadata serves as a critical link between raw genetic data and useful scientific insights, giving necessary context and allowing researchers to draw practical findings from their investigations. Sample validation approaches improve the reliability and reproducibility of the results, providing users confidence in the quality of the genetic data provided by ERGA. Looking ahead, ERGA envisions its decentralized infrastructure serving as a model for global collaborative research efforts. By embracing diversity, encouraging cooperation, and pushing for open access to data and resources, ERGA hopes to catalyze scientific discovery and generate positive change in the field of biodiversity genomics. ERGA aims to promote a more equitable and sustainable future for all by ongoing interaction with stakeholders, intensive outreach and education activities, and policy change advocacy. In addition to its immediate goals, ERGA considers the long-term implications of its work. As genomic technology progresses, the potential application of high-quality reference genomes will continue to grow. From informing conservation efforts and illuminating evolutionary histories to revolutionizing healthcare and agriculture, it is likely that ERGA's contributions will have far-reaching consequences for people and the planet as a whole. Furthermore, ERGA understands the importance of interdisciplinary collaboration in addressing the difficult challenges of the twenty-first century. ERGA aims to integrate genetic research into larger initiatives to promote sustainability and biodiversity conservation by forming relationships with stakeholders from other areas, such as policymakers, conservationists, and indigenous groups. Through shared knowledge and community action, ERGA seeks to create a future in which mankind coexists peacefully with the natural world, guided by a thorough grasp of its genetic legacy and ecological interconnectivity. Finally, the manuscript exemplifies ERGA's collaborative ambitions and achievements, capturing the spirit of creativity and collaboration that defines this ground-breaking effort. As ERGA continues to push the boundaries of genetic research, it remains dedicated to scientific excellence, inclusivity, and the quest of knowledge for the benefit of society. I wholeheartedly recommend the publication of this groundbreaking initiative, offering my enthusiastic endorsement for its valuable contribution to the scientific community. References Lewin, H. A., Robinson, G. E., Kress, W. J., Baker, W. J., Coddington, J., Crandall, K. A., Durbin, R., Edwards, S. V., Forest, F., Gilbert, M. T. P., Goldstein, M. M., Grigoriev, I. V., Hackett, K. J., Haussler, D., Jarvis, E. D., Johnson, W. E., Patrinos, A., Richards, S., Castilla-Rubio, J. C., … Zhang, G. (2018). Earth BioGenome Project: Sequencing life for the future of life. Proceedings of the National Academy of Sciences, 115(17), 4325–4333. https://doi.org/10.1073/pnas.1720115115 Mazzoni, C. J., Claudio, C.i, Waterhouse, R. M. (2023). Biodiversity: an atlas of European reference genomes. Nature 619 : 252-252. https://doi.org/10.1038/d41586-023-02229-w Mc Cartney, A. M., Formenti, G., Mouton, A., Panis, D. de, Marins, L. S., Leitão, H. G., Diedericks, G., Kirangwa, J., Morselli, M., Salces-Ortiz, J., Escudero, N., Iannucci, A., Natali, C., Svardal, H., Fernández, R., Pooter, T. de, Joris, G., Strazisar, M., Wood, J., … Mazzoni, C. J. (2024). The European Reference Genome Atlas: piloting a decentralised approach to equitable biodiversity genomics. bioRxiv, ver. 4 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2023.09.25.559365 | The European Reference Genome Atlas: piloting a decentralised approach to equitable biodiversity genomics | Ann M Mc Cartney, Giulio Formenti, Alice Mouton, Claudio Ciofi, Robert M Waterhouse, Camila J Mazzoni, Diego De Panis, Luisa S Schlude Marins, Henrique G Leitao, Genevieve Diedericks, Joseph Kirangwa, Marco Morselli, Judit Salces, Nuria Escudero, ... | <p>English: A global genome database of all of Earth's species diversity could be a treasure trove of scientific discoveries. However, regardless of the major advances in genome sequencing technologies, only a tiny fraction of species have genomic... | | Bioinformatics, ERGA Pilot | Jitendra Narayan | Justin Ideozu, Eric Crandall | 2023-10-01 01:03:58 | |
19 Jul 2021
TransPi - a comprehensive TRanscriptome ANalysiS PIpeline for de novo transcriptome assemblyTransPI: A balancing act between transcriptome assemblersRecommended by Oleg Simakov based on reviews by Gustavo Sanchez and Juan Daniel Montenegro CabreraEver since the introduction of the first widely usable assemblers for transcriptomic reads (Huang and Madan 1999; Schulz et al. 2012; Simpson et al. 2009; Trapnell et al. 2010, and many more), it has been a technical challenge to compare different methods and to choose the “right” or “best” assembly. It took years until the first widely accepted set of benchmarks beyond raw statistical evaluation became available (e.g., Parra, Bradnam, and Korf 2007; Simão et al. 2015). However, an approach to find the right balance between the number of transcripts or isoforms vs. evolutionary completeness measures has been lacking. This has been particularly pronounced in the field of non-model organisms (i.e., wild species that lack a genomic reference). Often, studies in this area employed only one set of assembly tools (the most often used to this day being Trinity, Haas et al. 2013; Grabherr et al. 2011). While it was relatively straightforward to obtain an initial assembly, its validation, annotation, as well its application to the particular purpose that the study was designed for (phylogenetics, differential gene expression, etc) lacked a clear workflow. This led to many studies using a custom set of tools with ensuing various degrees of reproducibility. TransPi (Rivera-Vicéns et al. 2021) fills this gap by first employing a meta approach using several available transcriptome assemblers and algorithms to produce a combined and reduced transcriptome assembly, then validating and annotating the resulting transcriptome. Notably, TransPI performs an extensive analysis/detection of chimeric transcripts, the results of which show that this new tool often produces fewer misassemblies compared to Trinity. TransPI not only generates a final report that includes the most important plots (in clickable/zoomable format) but also stores all relevant intermediate files, allowing advanced users to take a deeper look and/or experiment with different settings. As running TransPi is largely automated (including its installation via several popular package managers), it is very user-friendly and is likely to become the new "gold standard" for transcriptome analyses, especially of non-model organisms. References Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature Biotechnology, 29, 644–652. https://doi.org/10.1038/nbt.1883 Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, Couger MB, Eccles D, Li B, Lieber M, MacManes MD, Ott M, Orvis J, Pochet N, Strozzi F, Weeks N, Westerman R, William T, Dewey CN, Henschel R, LeDuc RD, Friedman N, Regev A (2013) De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nature Protocols, 8, 1494–1512. https://doi.org/10.1038/nprot.2013.084 Huang X, Madan A (1999) CAP3: A DNA Sequence Assembly Program. Genome Research, 9, 868–877. https://doi.org/10.1101/gr.9.9.868 Parra G, Bradnam K, Korf I (2007) CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics, 23, 1061–1067. https://doi.org/10.1093/bioinformatics/btm071 Rivera-Vicéns RE, Garcia-Escudero CA, Conci N, Eitel M, Wörheide G (2021) TransPi – a comprehensive TRanscriptome ANalysiS PIpeline for de novo transcriptome assembly. bioRxiv, 2021.02.18.431773, ver. 3 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2021.02.18.431773 Schulz MH, Zerbino DR, Vingron M, Birney E (2012) Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics, 28, 1086–1092. https://doi.org/10.1093/bioinformatics/bts094 Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM (2015) BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics, 31, 3210–3212. https://doi.org/10.1093/bioinformatics/btv351 Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJM, Birol İ (2009) ABySS: A parallel assembler for short read sequence data. Genome Research, 19, 1117–1123. https://doi.org/10.1101/gr.089532.108 Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L (2010) Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature Biotechnology, 28, 511–515. https://doi.org/10.1038/nbt.1621 | TransPi - a comprehensive TRanscriptome ANalysiS PIpeline for de novo transcriptome assembly | Ramon E Rivera-Vicens, Catalina Garcia-Escudero, Nicola Conci, Michael Eitel, Gert Wörheide | <p style="text-align: justify;">The use of RNA-Seq data and the generation of de novo transcriptome assemblies have been pivotal for studies in ecology and evolution. This is distinctly true for non-model organisms, where no genome information is ... | | Bioinformatics, Evolutionary genomics | Oleg Simakov | 2021-02-18 20:56:08 | ||
06 May 2022

A deep dive into genome assemblies of non-vertebrate animalsDiving, and even digging, into the wild jungle of annotation pathways for non-vertebrate animalsRecommended by Francois Sabot based on reviews by Yann Bourgeois, Cécile Monat, Valentina Peona and Benjamin Istace
In their paper, Guiglielmoni et al. propose we pick up our snorkels and palms and take "A deep dive into genome assemblies of non-vertebrate animals" (1). Indeed, while numerous assembly-related tools were developed and tested for human genomes (or at least vertebrates such as mice), very few were tested on non-vertebrate animals so far. Moreover, most of the benchmarks are aimed at raw assembly tools, and very few offer a guide from raw reads to an almost finished assembly, including quality control and phasing. This huge and exhaustive review starts with an overview of the current sequencing technologies, followed by the theory of the different approaches for assembly and their implementation. For each approach, the authors present some of the most representative tools, as well as the limits of the approach. The authors additionally present all the steps required to obtain an almost complete assembly at a chromosome-scale, with all the different technologies currently available for scaffolding, QC, and phasing, and the way these tools can be applied to non-vertebrates animals. Finally, they propose some useful advice on the choice of the different approaches (but not always tools, see below), and advocate for a robust genome database with all information on the way the assembly was obtained. This review is a very complete one for now and is a very good starting point for any student or scientist interested to start working on genome assembly, from either model or non-model organisms. However, the authors do not provide a list of tools or a benchmark of them as a recommendation. Why? Because such a proposal may be obsolete in less than a year.... Indeed, with the explosion of the 3rd generation of sequencing technology, assembly tools (from different steps) are constantly evolving, and their relative performance increases on a monthly basis. In addition, some tools are really efficient at the time of a review or of an article, but are not further developed later on, and thus will not evolve with the technology. We have all seen it with wonderful tools such as Chiron (2) or TopHat (3), which were very promising ones, but cannot be developed further due to the stop of the project, the end of the contract of the post-doc in charge of the development, or the decision of the developer to switch to another paradigm. Such advice would, therefore, need to be constantly updated. Thus, the manuscript from Guiglielmoni et al will be an almost intemporal one (up to the next sequencing revolution at last), and as they advocated for a more informed genome database, I think we should consider a rolling benchmarking system (tools, genome and sequence dataset) allowing to keep the performance of the tools up-to-date, and to propose the best set of assembly tools for a given type of genome. References 1. Guiglielmoni N, Rivera-Vicéns R, Koszul R, Flot J-F (2022) A Deep Dive into Genome Assemblies of Non-vertebrate Animals. Preprints, 2021110170, ver. 3 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.20944/preprints202111.0170 2. Teng H, Cao MD, Hall MB, Duarte T, Wang S, Coin LJM (2018) Chiron: translating nanopore raw signal directly into nucleotide sequence using deep learning. GigaScience, 7, giy037. https://doi.org/10.1093/gigascience/giy037 3. Trapnell C, Pachter L, Salzberg SL (2009) TopHat: discovering splice junctions with RNA-Seq. Bioinformatics, 25, 1105–1111. https://doi.org/10.1093/bioinformatics/btp120 | A deep dive into genome assemblies of non-vertebrate animals | Nadège Guiglielmoni, Ramón Rivera-Vicéns, Romain Koszul, Jean-François Flot | <p style="text-align: justify;">Non-vertebrate species represent about ∼95% of known metazoan (animal) diversity. They remain to this day relatively unexplored genetically, but understanding their genome structure and function is pivotal for expan... | | Bioinformatics, Evolutionary genomics | Francois Sabot | Valentina Peona, Benjamin Istace, Cécile Monat, Yann Bourgeois | 2021-11-10 17:47:31 | |
13 Jul 2022

Karyorelict ciliates use an ambiguous genetic code with context-dependent stop/sense codonsAn accident frozen in time: the ambiguous stop/sense genetic code of karyorelict ciliatesRecommended by Iker Irisarri based on reviews by Vittorio Boscaro and 2 anonymous reviewers
Several variations of the “universal” genetic code are known. Among the most striking are those where a codon can either encode for an amino acid or a stop signal depending on the context. Such ambiguous codes are known to have evolved in eukaryotes multiple times independently, particularly in ciliates – eight different codes have so far been discovered (1). We generally view such genetic codes are rare ‘variants’ of the standard code restricted to single species or strains, but this might as well reflect a lack of study of closely related species. In this study, Seah and co-authors (2) explore the possibility of codon reassignment in karyorelict ciliates closely related to Parduczia sp., which has been shown to contain an ambiguous genetic code (1). Here, single-cell transcriptomics are used, along with similar available data, to explore the possibility of codon reassignment across the diversity of Karyorelictea (four out of the six recognized families). Codon reassignments were inferred from their frequencies within conserved Pfam (3) protein domains, whereas stop codons were inferred from full-length transcripts with intact 3’-UTRs. Results show the reassignment of UAA and UAG stop codons to code for glutamine (Q) and the reassignment of the UGA stop codon into tryptophan (W). This occurs only within the coding sequences, whereas the end of transcription is marked by UGA as the main stop codon, and to a lesser extent by UAA. In agreement with a previous model proposed that explains the functioning of ambiguous codes (1,4), the authors observe a depletion of in-frame UGAs before the UGA codon that indicates the stop, thus avoiding premature termination of transcription. The inferred codon reassignments occur in all studied karyorelicts, including the previously studied Parduczia sp. Despite the overall clear picture, some questions remain. Data for two out of six main karyorelict lineages are so far absent and the available data for Cryptopharyngidae was inconclusive; the phylogenetic affinities of Cryptopharyngidae have also been questioned (5). This indicates the need for further study of this interesting group of organisms. As nicely discussed by the authors, experimental evidence could further strengthen the conclusions of this paper, including ribosome profiling, mass spectrometry – as done for Condylostoma (1) – or even direct genetic manipulation. The uniformity of the ambiguous genetic code across karyorelicts might at first seem dull, but when viewed in a phylogenetic context character distribution strongly suggest that this genetic code has an ancient origin in the karyorelict ancestor ~455 Ma in the Proterozoic (6). This ambiguous code is also not a rarity of some obscure species, but it is shared by ciliates that are very diverse and ecologically important. The origin of the karyorelict code is also intriguing. Adaptive arguments suggest that it could confer robustness to mutations causing premature stop codons. However, we lack evidence for ambiguous codes being linked to specific habitats of lifestyles that could account for it. Instead, the authors favor the neutral view of an ancient “frozen accident”, fixed stochastically simply because it did not pose a significant selective disadvantage. Once a stop codon is reassigned to an amino acid, it is increasingly difficult to revert this without the deleterious effect of prematurely terminating translation. At the end, the origin of the genetic code itself is thought to be a frozen accident too (7). References 1. Swart EC, Serra V, Petroni G, Nowacki M. Genetic codes with no dedicated stop codon: Context-dependent translation termination. Cell 2016;166: 691–702. https://doi.org/10.1016/j.cell.2016.06.020 2. Seah BKB, Singh A, Swart EC (2022) Karyorelict ciliates use an ambiguous genetic code with context-dependent stop/sense codons. bioRxiv, 2022.04.12.488043. ver. 4 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2022.04.12.488043 3. Mistry J, Chuguransky S, Williams L, Qureshi M, Salazar GA, Sonnhammer ELL, Tosatto SCE, Paladin L, Raj S, Richardson LJ, Finn RD, Bateman A. Pfam: The protein families database in 2021, Nuc Acids Res 2020;49: D412-D419. https://doi.org/10.1093/nar/gkaa913 4. Alkalaeva E, Mikhailova T. Reassigning stop codons via translation termination: How a few eukaryotes broke the dogma. Bioessays. 2017;39. https://doi.org/10.1002/bies.201600213 5. Xu Y, Li J, Song W, Warren A. Phylogeny and establishment of a new ciliate family, Wilbertomorphidae fam. nov. (Ciliophora, Karyorelictea), a highly specialized taxon represented by Wilbertomorpha colpoda gen. nov., spec. nov. J Eukaryot Microbiol. 2013;60: 480–489. https://doi.org/10.1111/jeu.12055 6. Fernandes NM, Schrago CG. A multigene timescale and diversification dynamics of Ciliophora evolution. Mol Phylogenet Evol. 2019;139: 106521. https://doi.org/10.1016/j.ympev.2019.106521 7. Crick FH. The origin of the genetic code. J Mol Biol. 1968;38: 367–379. https://doi.org/10.1016/0022-2836(68)90392-6 | Karyorelict ciliates use an ambiguous genetic code with context-dependent stop/sense codons | Brandon Kwee Boon Seah, Aditi Singh, Estienne Carl Swart | <p style="text-align: justify;">In ambiguous stop/sense genetic codes, the stop codon(s) not only terminate translation but can also encode amino acids. Such codes have evolved at least four times in eukaryotes, twice among ciliates (<em>Condylost... | | Bioinformatics, Evolutionary genomics | Iker Irisarri | 2022-05-02 11:06:10 |




