
Latest recommendations

| Id | Title * | Authors * | Abstract * ▲ | Picture * | Thematic fields * | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
15 Jan 2024
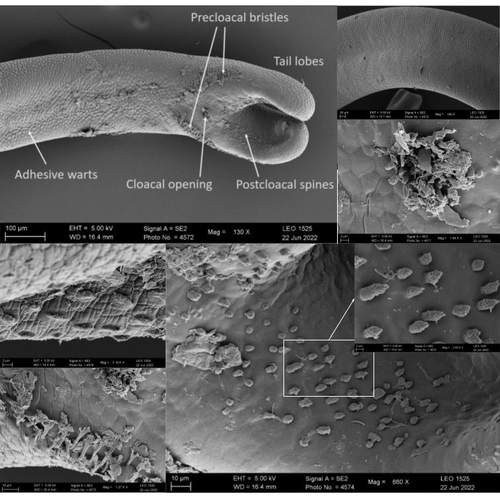
The genome sequence of the Montseny horsehair worm, Gordionus montsenyensis sp. nov., a key resource to investigate Ecdysozoa evolutionEmbarking on a novel journey in Metazoa evolution through the pioneering sequencing of a key underrepresented lineageRecommended by Juan C. Opazo based on reviews by Gonzalo Riadi and 2 anonymous reviewers based on reviews by Gonzalo Riadi and 2 anonymous reviewers
Whole genome sequences are revolutionizing our understanding across various biological fields. They not only shed light on the evolution of genetic material but also uncover the genetic basis of phenotypic diversity. The sequencing of underrepresented lineages, such as the one presented in this study, is of critical importance. It is crucial in filling significant gaps in our understanding of Metazoa evolution. Despite the wealth of genome sequences in public databases, it is crucial to acknowledge that some lineages across the Tree of Life are underrepresented or absent. This research represents a significant step towards addressing this imbalance, contributing to the collective knowledge of the global scientific community. In this genome note, as part of the European Reference Genome Atlas pilot effort to generate reference genomes for European biodiversity (Mc Cartney et al. 2023), Klara Eleftheriadi and colleagues (Eleftheriadi et al. 2023) make a significant effort to add a genome sequence of an unrepresented group in the animal Tree of Life. More specifically, they present a taxonomic description and chromosome-level genome assembly of a newly described species of horsehair worm (Gordionus montsenyensis). Their sequence methodology gave rise to an assembly of 396 scaffolds totaling 288 Mb, with an N50 value of 64.4 Mb, where 97% of this assembly is grouped into five pseudochromosomes. The nuclear genome annotation predicted 10,320 protein-coding genes, and they also assembled the circular mitochondrial genome into a 15-kilobase sequence. The selection of a species representing the phylum Nematomorpha, a group of parasitic organisms belonging to the Ecdysozoa lineage, is good, since today, there is only one publicly available genome for this animal phylum (Cunha et al. 2023). Interestingly, this article shows, among other things, that the species analyzed has lost ∼30% of the universal Metazoan genes. Efforts, like the one performed by Eleftheriadi and colleagues, are necessary to gain more insights, for example, on the evolution of this massive gene lost in this group of animals.
Cunha, T. J., de Medeiros, B. A. S, Lord, A., Sørensen, M. V., and Giribet, G. (2023). Rampant Loss of Universal Metazoan Genes Revealed by a Chromosome-Level Genome Assembly of the Parasitic Nematomorpha. Current Biology, 33 (16): 3514–21.e4. https://doi.org/10.1016/j.cub.2023.07.003 Eleftheriadi, K., Guiglielmoni, N., Salces-Ortiz, J., Vargas-Chavez, C., Martínez-Redondo, G. I., Gut, M., Flot, J.-F., Schmidt-Rhaesa, A., and Fernández, R. (2023). The Genome Sequence of the Montseny Horsehair worm, Gordionus montsenyensis sp. Nov., a Key Resource to Investigate Ecdysozoa Evolution. bioRxiv, ver. 3 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2023.06.26.546503 Mc Cartney, A. M., Formenti, G., Mouton, A., De Panis, D., Marins, L. S., Leitão, H. G., Diedericks, G., et al. (2023). The European Reference Genome Atlas: Piloting a Decentralised Approach to Equitable Biodiversity Genomics. bioRxiv. https://doi.org/10.1101/2023.09.25.559365 | The genome sequence of the Montseny horsehair worm, *Gordionus montsenyensis* sp. nov., a key resource to investigate Ecdysozoa evolution | Eleftheriadi Klara, Guiglielmoni Nadège, Salces-Ortiz Judit, Vargas-Chávez Carlos, Martínez-Redondo Gemma I, Gut Marta, Flot Jean François, Schmidt-Rhaesa Andreas, Fernández Rosa | <p>Nematomorpha, also known as Gordiacea or Gordian worms, are a phylum of parasitic organisms that belong to the Ecdysozoa, a clade of invertebrate animals characterized by molting. They are one of the less scientifically studied animal phyla, an... | | ERGA Pilot | Juan C. Opazo | 2023-06-29 10:31:36 | ||
09 Aug 2023
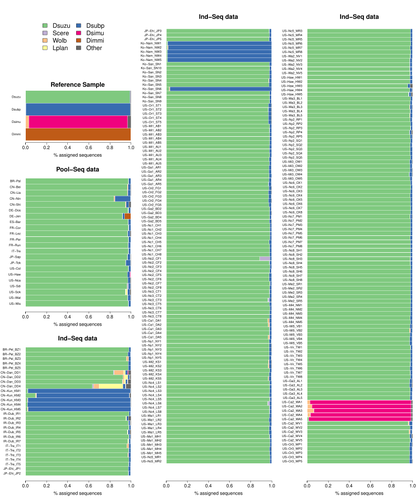
Efficient k-mer based curation of raw sequence data: application in Drosophila suzukiiDecontaminating reads, not contigsRecommended by Nicolas Galtier based on reviews by Marie Cariou and Denis BaurainContamination, the presence of foreign DNA sequences in a sample of interest, is currently a major problem in genomics. Because contamination is often unavoidable at the experimental stage, it is increasingly recognized that the processing of high-throughput sequencing data must include a decontamination step. This is usually performed after the many sequence reads have been assembled into a relatively small number of contigs. Dubious contigs are then discarded based on their composition (e.g. GC-content) or because they are highly similar to a known piece of DNA from a foreign species. Here [1], Mathieu Gautier explores a novel strategy consisting in decontaminating reads, not contigs. Why is this promising? Assembly programs and algorithms are complex, and it is not easy to predict, or monitor, how they handle contaminant reads. Ideally, contaminant reads will be assembled into obvious contaminant contigs. However, there might be more complex situations, such as chimeric contigs with alternating genuine and contaminant segments. Decontaminating at the read level, if possible, should eliminate such unfavorable situations where sequence information from contaminant and target samples are intimately intertwined by an assembler. To achieve this aim, Gautier proposes to use methods initially designed for the analysis of metagenomic data. This is pertinent since the decontamination process involves considering a sample as a mixture of different sources of DNA. The programs used here, CLARK and CLARK-L, are based on so-called k-mer analysis, meaning that the similarity between a read to annotate and a reference sequence is measured by how many sub-sequences (of length 31 base pairs for CLARK and 27 base pairs for CLARK-L) they share. This is notoriously more efficient than traditional sequence alignment algorithms when it comes to comparing a very large number of (most often unrelated) sequences. This is, therefore, a reference-based approach, in which the reads from a sample are assigned to previously sequenced genomes based on k-mer content. This original approach is here specifically applied to the case of Drosophila suzukii, an invasive pest damaging fruit production in Europe and America. Fortunately, Drosophila is a genus of insects with abundant genomic resources, including high-quality reference genomes in dozens of species. Having calibrated and validated his pipeline using data sets of known origins, Gautier quantifies in each of 258 presumed D. suzukii samples the proportion of reads that likely belong to other species of fruit flies, or to fruit fly-associated microbes. This proportion is close to one in 16 samples, which clearly correspond to mis-labelled individuals. It is non-negligible in another ~10 samples, which really correspond to D. suzukii individuals. Most of these reads of unexpected origin are contaminants and should be filtered out. Interestingly, one D. suzukii sample contains a substantial proportion of reads from the closely related D. subpulchera, which might instead reflect a recent episode of gene flow between these two species. The approach, therefore, not only serves as a crucial technical step, but also has the potential to reveal biological processes. Gautier's thorough, well-documented work will clearly benefit the ongoing and future research on D. suzuki, and Drosophila genomics in general. The author and reviewers rightfully note that, like any reference-based approach, this method is heavily dependent on the availability and quality of reference genomes - Drosophila being a favorable case. Building the reference database is a key step, and the interpretation of the output can only be made in the light of its content and gaps, as illustrated by Gautier's careful and detailed discussion of his numerous results. This pioneering study is a striking demonstration of the potential of metagenomic methods for the decontamination of high-throughput sequence data at the read level. The pipeline requires remarkably few computing resources, ensuring low carbon emission. I am looking forward to seeing it applied to a wide range of taxa and samples.
Reference [1] Gautier Mathieu. Efficient k-mer based curation of raw sequence data: application in Drosophila suzukii. bioRxiv, 2023.04.18.537389, ver. 2, peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2023.04.18.537389 | Efficient k-mer based curation of raw sequence data: application in *Drosophila suzukii* | Gautier Mathieu | <p>Several studies have highlighted the presence of contaminated entries in public sequence repositories, calling for special attention to the associated metadata. Here, we propose and evaluate a fast and efficient kmer-based approach to assess th... | | Bioinformatics, Population genomics | Nicolas Galtier | 2023-04-20 22:05:13 | ||
11 Mar 2021

Gut microbial ecology of Xenopus tadpoles across life stagesA comprehensive look at Xenopus gut microbiota: effects of feed, developmental stages and parental transmissionRecommended by Wirulda Pootakham based on reviews by Vanessa Marcelino and 1 anonymous reviewerIt is well established that the gut microbiota play an important role in the overall health of their hosts (Jandhyala et al. 2015). To date, there are still a limited number of studies on the complex microbial communites inhabiting vertebrate digestive systems, especially the ones that also explored the functional diversity of the microbial community (Bletz et al. 2016). This preprint by Scalvenzi et al. (2021) reports a comprehensive study on the phylogenetic and metabolic profiles of the Xenopus gut microbiota. The author describes significant changes in the gut microbiome communities at different developmental stages and demonstrates different microbial community composition across organs. In addition, the study also investigates the impact of diet on the Xenopus tadpole gut microbiome communities as well as how the bacterial communities are transmitted from parents to the next generation. This is one of the first studies that addresses the interactions between gut bacteria and tadpoles during the development. The authors observe the dynamics of gut microbiome communities during tadpole growth and metamorphosis. They also explore host-gut microbial community metabolic interactions and demostrate the capacity of the microbiome to complement the metabolic pathways of the Xenopus genome. Although this study is limited by the use of Xenopus tadpoles in a laboratory, which are probably different from those in nature, I believe it still provides important and valuable information for the research community working on vertebrate’s microbiota and their interaction with the host. References Bletz et al. (2016). Amphibian gut microbiota shifts differentially in community structure but converges on habitat-specific predicted functions. Nature Communications, 7(1), 1-12. doi: https://doi.org/10.1038/ncomms13699 Jandhyala, S. M., Talukdar, R., Subramanyam, C., Vuyyuru, H., Sasikala, M., & Reddy, D. N. (2015). Role of the normal gut microbiota. World journal of gastroenterology: WJG, 21(29), 8787. doi: https://dx.doi.org/10.3748%2Fwjg.v21.i29.8787 Scalvenzi, T., Clavereau, I., Bourge, M. & Pollet, N. (2021) Gut microbial ecology of Xenopus tadpoles across life stages. bioRxiv, 2020.05.25.110734, ver. 4 peer-reviewed and recommended by Peer community in Geonmics. https://doi.org/10.1101/2020.05.25.110734 | Gut microbial ecology of Xenopus tadpoles across life stages | Thibault Scalvenzi, Isabelle Clavereau, Mickael Bourge, Nicolas Pollet | <p><strong>Background</strong> The microorganism world living in amphibians is still largely under-represented and under-studied in the literature. Among anuran amphibians, African clawed frogs of the Xenopus genus stand as well-characterized mode... | | Evolutionary genomics, Metagenomics, Vertebrates | Wirulda Pootakham | 2020-05-25 14:01:19 | ||
08 Nov 2022

Somatic mutation detection: a critical evaluation through simulations and reanalyses in oaksHow to best call the somatic mosaic tree?Recommended by Nicolas Bierne based on reviews by 2 anonymous reviewersAny multicellular organism is a molecular mosaic with some somatic mutations accumulated between cell lineages. Big long-lived trees have nourished this imaginary of a somatic mosaic tree, from the observation of spectacular phenotypic mosaics and also because somatic mutations are expected to potentially be passed on to gametes in plants (review in Schoen and Schultz 2019). The lower cost of genome sequencing now offers the opportunity to tackle the issue and identify somatic mutations in trees. However, when it comes to characterizing this somatic mosaic from genome sequences, things become much more difficult than one would think in the first place. What separates cell lineages ontogenetically, in cell division number, or in time? How to sample clonal cell populations? How do somatic mutations distribute in a population of cells in an organ or an organ sample? Should they be fixed heterozygotes in the sample of cells sequenced or be polymorphic? Do we indeed expect somatic mutations to be fixed? How should we identify and count somatic mutations? To date, the detection of somatic mutations has mostly been done with a single variant caller in a given study, and we have little perspective on how different callers provide similar or different results. Some studies have used standard SNP callers that assumed a somatic mutation is fixed at the heterozygous state in the sample of cells, with an expected allele coverage ratio of 0.5, and less have used cancer callers, designed to detect mutations in a fraction of the cells in the sample. However, standard SNP callers detect mutations that deviate from a balanced allelic coverage, and different cancer callers can have different characteristics that should affect their outcomes. In order to tackle these issues, Schmitt et al. (2022) conducted an extensive simulation analysis to compare different variant callers. Then, they reanalyzed two large published datasets on pedunculate oak, Quercus robur. The analysis of in silico somatic mutations allowed the authors to evaluate the performance of different variant callers as a function of the allelic fraction of somatic mutations and the sequencing depth. They found one of the seven callers to provide better and more robust calls for a broad set of allelic fractions and sequencing depths. The reanalysis of published datasets in oaks with the most effective cancer caller of the in silico analysis allowed them to identify numerous low-frequency mutations that were missed in the original studies. I recommend the study of Schmitt et al. (2022) first because it shows the benefit of using cancer callers in the study of somatic mutations, whatever the allelic fraction you are interested in at the end. You can select fixed heterozygotes if this is your ultimate target, but cancer callers allow you to have in addition a valuable overview of the allelic fractions of somatic mutations in your sample, and most do as well as SNP callers for fixed heterozygous mutations. In addition, Schmitt et al. (2022) provide the pipelines that allow investigating in silico data that should correspond to a given study design, encouraging to compare different variant callers rather than arbitrarily going with only one. We can anticipate that the study of somatic mutations in non-model species will increasingly attract attention now that multiple tissues of the same individual can be sequenced at low cost, and the study of Schmitt et al. (2022) paves the way for questioning and choosing the best variant caller for the question one wants to address. References Schoen DJ, Schultz ST (2019) Somatic Mutation and Evolution in Plants. Annual Review of Ecology, Evolution, and Systematics, 50, 49–73. https://doi.org/10.1146/annurev-ecolsys-110218-024955 Schmitt S, Leroy T, Heuertz M, Tysklind N (2022) Somatic mutation detection: a critical evaluation through simulations and reanalyses in oaks. bioRxiv, 2021.10.11.462798. ver. 4 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2021.10.11.462798 | Somatic mutation detection: a critical evaluation through simulations and reanalyses in oaks | Sylvain Schmitt, Thibault Leroy, Myriam Heuertz, Niklas Tysklind | <p style="text-align: justify;">1. Mutation, the source of genetic diversity, is the raw material of evolution; however, the mutation process remains understudied, especially in plants. Using both a simulation and reanalysis framework, we set out ... | | Bioinformatics, Plants | Nicolas Bierne | Anonymous, Anonymous | 2022-04-28 13:24:19 | |
27 Apr 2021

Uncovering transposable element variants and their potential adaptive impact in urban populations of the malaria vector Anopheles coluzziiAnopheles coluzzii, a new system to study how transposable elements may foster adaptation to urban environmentsRecommended by Anne Roulin based on reviews by Yann Bourgeois and 1 anonymous reviewerTransposable elements (TEs) are mobile DNA sequences that can increase their copy number and move from one location to another within the genome [1]. Because of their transposition dynamics, TEs constitute a significant fraction of eukaryotic genomes. TEs are also known to play an important functional role and a wealth of studies has now reported how TEs may influence single host traits [e.g. 2–4]. Given that TEs are more likely than classical point mutations to cause extreme changes in gene expression and phenotypes, they might therefore be especially prone to produce the raw diversity necessary for individuals to respond to challenging environments [5,6] such as the ones found in urban area.
| Uncovering transposable element variants and their potential adaptive impact in urban populations of the malaria vector Anopheles coluzzii | Carlos Vargas-Chavez, Neil Michel Longo Pendy, Sandrine E. Nsango, Laura Aguilera, Diego Ayala, and Josefa González | <p style="text-align: justify;">Background</p> <p style="text-align: justify;">Anopheles coluzzii is one of the primary vectors of human malaria in sub-Saharan Africa. Recently, it has colonized the main cities of Central Africa threatening vecto... | | Evolutionary genomics | Anne Roulin | 2020-12-02 14:58:47 | ||
23 Mar 2022
Chromosomal rearrangements with stable repertoires of genes and transposable elements in an invasive forest-pathogenic fungusComparative genomics in the chestnut blight fungus Cryphonectria parasitica reveals large chromosomal rearrangements and a stable genome organizationRecommended by Sebastien Duplessis based on reviews by Benjamin Schwessinger and 1 anonymous reviewerAbout twenty-five years after the sequencing of the first fungal genome and a dozen years after the first plant pathogenic fungi genomes were sequenced, unprecedented international efforts have led to an impressive collection of genomes available for the community of mycologists in international databases (Goffeau et al. 1996, Dean et al. 2005; Spatafora et al. 2017). For instance, to date, the Joint Genome Institute Mycocosm database has collected more than 2,100 fungal genomes over the fungal tree of life (https://mycocosm.jgi.doe.gov). Such resources are paving the way for comparative genomics, population genomics and phylogenomics to address a large panel of questions regarding the biology and the ecology of fungal species. Early on, population genomics applied to pathogenic fungi revealed a great diversity of genome content and organization and a wide variety of variants and rearrangements (Raffaele and Kamoun 2012, Hartmann 2022). Such plasticity raises questions about how to choose a representative genome to serve as an ideal reference to address pertinent biological questions. Cryphonectria parasitica is a fungal pathogen that is infamous for the devastation of chestnut forests in North America after its accidental introduction more than a century ago (Anagnostakis 1987). Since then, it has been a quarantine species under surveillance in various parts of the world. As for other fungi causing diseases on forest trees, the study of adaptation to its host in the forest ecosystem and of its reproduction and dissemination modes is more complex than for crop-targeting pathogens. A first reference genome was published in 2020 for the chestnut blight fungus C. parasitica strain EP155 in the frame of an international project with the DOE JGI (Crouch et al. 2020). Another genome was then sequenced from the French isolate YVO003, which showed a few differences in the assembly suggesting possible rearrangements (Demené et al. 2019). Here the sequencing of a third isolate ESM015 from the native area of C. parasitica in Japan allows to draw broader comparative analysis and particularly to compare between native and introduced isolates (Demené et al. 2022). Demené and collaborators report on a new genome sequence using up-to-date long-read sequencing technologies and they provide an improved genome assembly. Comparison with previously published C. parasitica genomes did not reveal dramatic changes in the overall chromosomal landscapes, but large rearrangements could be spotted. Despite these rearrangements, the genome content and organization – i.e. genes and repeats – remain stable, with a limited number of genes gains and losses. As in any fungal plant pathogen genome, the repertoire of candidate effectors predicted among secreted proteins was more particularly scrutinized. Such effector genes have previously been reported in other pathogens in repeat-enriched plastic genomic regions with accelerated evolutionary rates under the pressure of the host immune system (Raffaele and Kamoun 2012). Demené and collaborators established a list of priority candidate effectors in the C. parasitica gene catalog likely involved in the interaction with the host plant which will require more attention in future functional studies. Six major inter-chromosomal translocations were detected and are likely associated with double break strands repairs. The authors speculate on the possible effects that these translocations may have on gene organization and expression regulation leading to dramatic phenotypic changes in relation to introduction and invasion in new continents and the impact regarding sexual reproduction in this fungus (Demené et al. 2022). I recommend this article not only because it is providing an improved assembly of a reference genome for C. parasitica, but also because it adds diversity in terms of genome references availability, with a third high-quality assembly. Such an effort in the tree pathology community for a pathogen under surveillance is of particular importance for future progress in post-genomic analysis, e.g. in further genomic population studies (Hartmann 2022). References Anagnostakis SL (1987) Chestnut Blight: The Classical Problem of an Introduced Pathogen. Mycologia, 79, 23–37. https://doi.org/10.2307/3807741 Crouch JA, Dawe A, Aerts A, Barry K, Churchill ACL, Grimwood J, Hillman BI, Milgroom MG, Pangilinan J, Smith M, Salamov A, Schmutz J, Yadav JS, Grigoriev IV, Nuss DL (2020) Genome Sequence of the Chestnut Blight Fungus Cryphonectria parasitica EP155: A Fundamental Resource for an Archetypical Invasive Plant Pathogen. Phytopathology®, 110, 1180–1188. https://doi.org/10.1094/PHYTO-12-19-0478-A Dean RA, Talbot NJ, Ebbole DJ, Farman ML, Mitchell TK, Orbach MJ, Thon M, Kulkarni R, Xu J-R, Pan H, Read ND, Lee Y-H, Carbone I, Brown D, Oh YY, Donofrio N, Jeong JS, Soanes DM, Djonovic S, Kolomiets E, Rehmeyer C, Li W, Harding M, Kim S, Lebrun M-H, Bohnert H, Coughlan S, Butler J, Calvo S, Ma L-J, Nicol R, Purcell S, Nusbaum C, Galagan JE, Birren BW (2005) The genome sequence of the rice blast fungus Magnaporthe grisea. Nature, 434, 980–986. https://doi.org/10.1038/nature03449 Demené A., Laurent B., Cros-Arteil S., Boury C. and Dutech C. 2022. Chromosomal rearrangements with stable repertoires of genes and transposable elements in an invasive forest-pathogenic fungus. bioRxiv, 2021.03.09.434572, ver.6 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2021.03.09.434572 Goffeau A, Barrell BG, Bussey H, Davis RW, Dujon B, Feldmann H, Galibert F, Hoheisel JD, Jacq C, Johnston M, Louis EJ, Mewes HW, Murakami Y, Philippsen P, Tettelin H, Oliver SG (1996) Life with 6000 Genes. Science, 274, 546–567. https://doi.org/10.1126/science.274.5287.546 Hartmann FE (2022) Using structural variants to understand the ecological and evolutionary dynamics of fungal plant pathogens. New Phytologist, 234, 43–49. https://doi.org/10.1111/nph.17907 Raffaele S, Kamoun S (2012) Genome evolution in filamentous plant pathogens: why bigger can be better. Nature Reviews Microbiology, 10, 417–430. https://doi.org/10.1038/nrmicro2790 Spatafora JW, Aime MC, Grigoriev IV, Martin F, Stajich JE, Blackwell M (2017) The Fungal Tree of Life: from Molecular Systematics to Genome-Scale Phylogenies. Microbiology Spectrum, 5, 5.5.03. https://doi.org/10.1128/microbiolspec.FUNK-0053-2016 | Chromosomal rearrangements with stable repertoires of genes and transposable elements in an invasive forest-pathogenic fungus | Arthur Demene, Benoit Laurent, Sandrine Cros-Arteil, Christophe Boury, Cyril Dutech | <p style="text-align: justify;">Chromosomal rearrangements have been largely described among eukaryotes, and may have important consequences on evolution of species. High genome plasticity has been often reported in Fungi, which may explain their ... | | Evolutionary genomics, Fungi | Sebastien Duplessis | 2021-03-12 14:18:20 | ||
07 Sep 2023
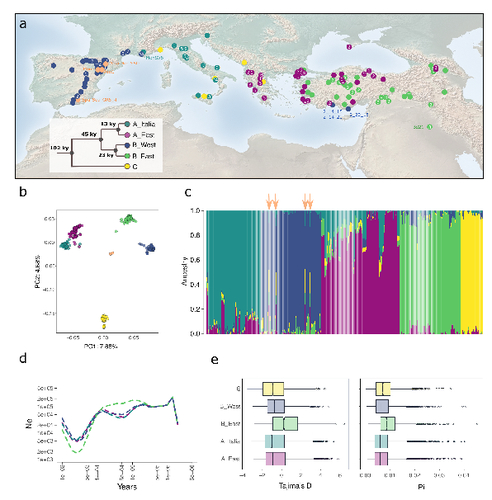
The demographic history of the wild crop relative Brachypodium distachyon is shaped by distinct past and present ecological nichesNatural variation and adaptation in Brachypodium distachyonRecommended by Josep Casacuberta based on reviews by Thibault Leroy and 1 anonymous reviewerIdentifying the genetic factors that allow plant adaptation is a major scientific question that is particularly relevant in the face of the climate change that we are already experiencing. To address this, it is essential to have genetic information on a high number of accessions (i.e., plants registered with unique accession numbers) growing under contrasting environmental conditions. There is already an important number of studies addressing these issues in the plant Arabidopsis thaliana, but there is a need to expand these analyses to species that play key roles in wild ecosystems and are close to very relevant crops, as is the case of grasses. The work of Minadakis, Roulin and co-workers (1) presents a Brachypodium distachyon panel of 332 fully sequences accessions that covers the whole species distribution across a wide range of bioclimatic conditions, which will be an invaluable tool to fill this gap. In addition, the authors use this data to start analyzing the population structure and demographic history of this plant, suggesting that the species experienced a shift of its distribution following the Last Glacial Maximum, which may have forced the species into new habitats. The authors also present a modeling of the niches occupied by B. distachyon together with an analysis of the genetic clades found in each of them, and start analyzing the different adaptive loci that may have allowed the species’ expansion into different bioclimatic areas. In addition to the importance of the resources made available by the authors for the scientific community, the analyses presented are well done and carefully discussed, and they highlight the potential of these new resources to investigate the genetic bases of plant adaptation. References 1. Nikolaos Minadakis, Hefin Williams, Robert Horvath, Danka Caković, Christoph Stritt, Michael Thieme, Yann Bourgeois, Anne C. Roulin. The demographic history of the wild crop relative Brachypodium distachyon is shaped by distinct past and present ecological niches. bioRxiv, 2023.06.01.543285, ver. 5 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2023.06.01.543285 | The demographic history of the wild crop relative *Brachypodium distachyon* is shaped by distinct past and present ecological niches | Nikolaos Minadakis, Hefin Williams, Robert Horvath, Danka Caković, Christoph Stritt, Michael Thieme, Yann Bourgeois, Anne C. Roulin | <p style="text-align: justify;">Closely related to economically important crops, the grass <em>Brachypodium distachyon</em> has been originally established as a pivotal species for grass genomics but more recently flourished as a model for develop... | | Evolutionary genomics, Functional genomics, Plants, Population genomics | Josep Casacuberta | 2023-06-14 15:28:30 | ||
07 Feb 2023
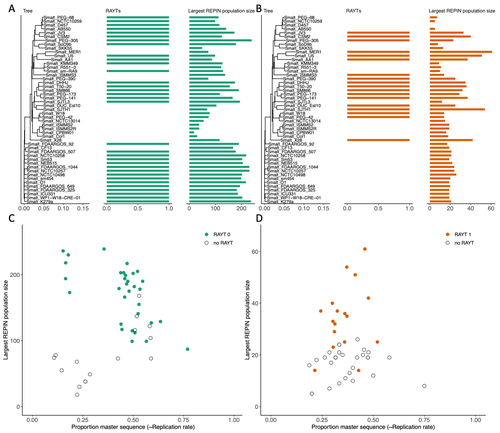
RAREFAN: A webservice to identify REPINs and RAYTs in bacterial genomesA workflow for studying enigmatic non-autonomous transposable elements across bacteriaRecommended by Gavin Douglas based on reviews by Sophie Abby and 1 anonymous reviewer
Repetitive extragenic palindromic sequences (REPs) are common repetitive elements in bacterial genomes (Gilson et al., 1984; Stern et al., 1984). In 2011, Bertels and Rainey identified that REPs are overrepresented in pairs of inverted repeats, which likely form hairpin structures, that they referred to as “REP doublets forming hairpins” (REPINs). Based on bioinformatics analyses, they argued that REPINs are likely selfish elements that evolved from REPs flanking particular transposes (Bertels and Rainey, 2011). These transposases, so-called REP-associated tyrosine transposases (RAYTs), were known to be highly associated with the REP content in a genome and to have characteristic upstream and downstream flanking REPs (Nunvar et al., 2010). The flanking REPs likely enable RAYT transposition, and their horizontal replication is physically linked to this process. In contrast, Bertels and Rainey hypothesized that REPINs are selfish elements that are highly replicated due to the similarity in arrangement to these RAYT-flanking REPs, but independent of RAYT transposition and generally with no impact on bacterial fitness (Bertels and Rainey, 2011). This last point was especially contentious, as REPINs are highly conserved within species (Bertels and Rainey, 2023), which is unusual for non-beneficial bacterial DNA (Mira et al., 2001). Bertels and Rainey have since refined their argument to be that REPINs must provide benefits to host cells, but that there are nonetheless signatures of intragenomic conflict in genomes associated with these elements (Bertels and Rainey, 2023). These signatures reflect the divergent levels of selections driving REPIN distribution: selection at the level of each DNA element and selection on each individual bacterium. I found this observation particularly interesting as I and my colleague recently argued that these divergent levels of selection, and the interaction between them, is key to understanding bacterial pangenome diversity (Douglas and Shapiro, 2021). REPINs could be an excellent system for investigating these levels of selection across bacteria more generally. The problem is that REPINs have not been widely characterized in bacterial genomes, partially because no bioinformatic workflow has been available for this purpose. To address this problem, Fortmann-Grote et al. (2023) developed RAREFAN, which is a web server for identifying RAYTs and associated REPINs in a set of input genomes. The authors showcase their tool by applying it to 49 Stenotrophomonas maltophilia genomes and providing examples of how to identify and assess RAYT-REPIN hits. The workflow requires several manual steps, but nonetheless represents a straightforward and standardized approach. Overall, this workflow should enable RAYTs and REPINs to be identified across diverse bacterial species, which will facilitate further investigation into the mechanisms driving their maintenance and spread. References Bertels F, Rainey PB (2023) Ancient Darwinian replicators nested within eubacterial genomes. BioEssays, 45, 2200085. https://doi.org/10.1002/bies.202200085 Bertels F, Rainey PB (2011) Within-Genome Evolution of REPINs: a New Family of Miniature Mobile DNA in Bacteria. PLOS Genetics, 7, e1002132. https://doi.org/10.1371/journal.pgen.1002132 Douglas GM, Shapiro BJ (2021) Genic Selection Within Prokaryotic Pangenomes. Genome Biology and Evolution, 13, evab234. https://doi.org/10.1093/gbe/evab234 Fortmann-Grote C, Irmer J von, Bertels F (2023) RAREFAN: A webservice to identify REPINs and RAYTs in bacterial genomes. bioRxiv, 2022.05.22.493013, ver. 4 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2022.05.22.493013 Gilson E, Clément J m., Brutlag D, Hofnung M (1984) A family of dispersed repetitive extragenic palindromic DNA sequences in E. coli. The EMBO Journal, 3, 1417–1421. https://doi.org/10.1002/j.1460-2075.1984.tb01986.x Mira A, Ochman H, Moran NA (2001) Deletional bias and the evolution of bacterial genomes. Trends in Genetics, 17, 589–596. https://doi.org/10.1016/S0168-9525(01)02447-7 Nunvar J, Huckova T, Licha I (2010) Identification and characterization of repetitive extragenic palindromes (REP)-associated tyrosine transposases: implications for REP evolution and dynamics in bacterial genomes. BMC Genomics, 11, 44. https://doi.org/10.1186/1471-2164-11-44 Stern MJ, Ames GF-L, Smith NH, Clare Robinson E, Higgins CF (1984) Repetitive extragenic palindromic sequences: A major component of the bacterial genome. Cell, 37, 1015–1026. https://doi.org/10.1016/0092-8674(84)90436-7 | RAREFAN: A webservice to identify REPINs and RAYTs in bacterial genomes | Frederic Bertels, Julia von Irmer, Carsten Fortmann-Grote | <p style="text-align: justify;">Compared to eukaryotes, repetitive sequences are rare in bacterial genomes and usually do not persist for long. Yet, there is at least one class of persistent prokaryotic mobile genetic elements: REPINs. REPINs are ... | | Bacteria and archaea, Bioinformatics, Evolutionary genomics, Viruses and transposable elements | Gavin Douglas | 2022-06-07 08:21:34 | ||
24 Feb 2023

MacSyFinder v2: Improved modelling and search engine to identify molecular systems in genomesA unique and customizable approach for functionally annotating prokaryotic genomesRecommended by Gavin Douglas based on reviews by Kwee Boon Brandon Seah and Max Emil Schön
Macromolecular System Finder (MacSyFinder) v2 (Néron et al., 2023) is a newly updated approach for performing functional annotation of prokaryotic genomes (Abby et al., 2014). This tool parses an input file of protein sequences from a single genome (either ordered by genome location or unordered) and identifies the presence of specific cellular functions (referred to as “systems”). These systems are called based on two criteria: (1) that the "quorum" of a minimum set of core proteins involved is reached the “quorum” of a minimum set of core proteins being involved that are present, and (2) that the genes encoding these proteins are in the expected genomic organization (e.g., within the same order in an operon), when ordered data is provided. I believe the MacSyFinder approach represents an improvement over more commonly used methods exactly because it can incorporate such information on genomic organization, and also because it is more customizable. Before properly appreciating these points, it is worth noting the norms and key challenges surrounding high-throughput functional annotation of prokaryotic genomes. Genome sequences are being added to online repositories at increasing rates, which has led to an enormous amount of bacterial genome diversity available to investigate (Altermann et al., 2022). A key aspect of understanding this diversity is the functional annotation step, which enables genes to be grouped into more biologically interpretable categories. For instance, gene calls can be mapped against existing Clusters of Orthologous Genes, which are themselves grouped into general categories such as ‘Transcription’ and ‘Lipid metabolism’ (Galperin et al., 2021). This approach is valuable but is primarily used for global summaries of functional annotations within a genome: for example, it could be useful to know that a genome is particularly enriched for genes involved in lipid metabolism. However, knowing that a particular gene is involved in the general process of lipid metabolism is less likely to be actionable. In other words, the desired specificity of a gene’s functional annotation will depend on the exact question being investigated. There is no shortage of functional ontologies in genomics that can be applied for this purpose (Douglas and Langille, 2021), and researchers are often overwhelmed by the choice of which functional ontology to use. In this context, giving researchers the ability to precisely specify the gene families and operon structures they are interested in identifying across genomes provides useful control over what precise functions they are profiling. Of course, most researchers will lack the information and/or expertise to fully take advantage of MacSyFinder’s customizable features, but having this option for specialized purposes is valuable. The other MacSyFinder feature that I find especially noteworthy is that it can incorporate genomic organization (e.g., of genes ordered in operons) when calling systems. This is a rare feature among commonly used tools for functional annotation and likely results in much higher specificity. As the authors note, this capability makes the co-occurrence of paralogs, and other divergent genes that share sequence similarity, to contribute less noise (i.e., they result in fewer false positive calls). It is important to emphasize that these features are not new additions in MacSyFinder v2, but there are many other valuable changes. Most practically, this release is written in Python 3, rather than the obsolete Python 2.7, and was made more computationally efficient, which will enable MacSyFinder to be more widely used and more easily maintained moving forward. In addition, the search algorithm for analyzing individual proteins was fundamentally updated as well. The authors show that their improvements to the search algorithm result in an 8% and 20% increase in the number of identified calls for single and multi-locus secretion systems, respectively. Taken together, MacSyFinder v2 represents both practical and scientific improvements over the previous version, which will be of great value to the field. References Abby SS, Néron B, Ménager H, Touchon M, Rocha EPC (2014) MacSyFinder: A Program to Mine Genomes for Molecular Systems with an Application to CRISPR-Cas Systems. PLOS ONE, 9, e110726. https://doi.org/10.1371/journal.pone.0110726 Altermann E, Tegetmeyer HE, Chanyi RM (2022) The evolution of bacterial genome assemblies - where do we need to go next? Microbiome Research Reports, 1, 15. https://doi.org/10.20517/mrr.2022.02 Douglas GM, Langille MGI (2021) A primer and discussion on DNA-based microbiome data and related bioinformatics analyses. Peer Community Journal, 1. https://doi.org/10.24072/pcjournal.2 Galperin MY, Wolf YI, Makarova KS, Vera Alvarez R, Landsman D, Koonin EV (2021) COG database update: focus on microbial diversity, model organisms, and widespread pathogens. Nucleic Acids Research, 49, D274–D281. https://doi.org/10.1093/nar/gkaa1018 Néron B, Denise R, Coluzzi C, Touchon M, Rocha EPC, Abby SS (2023) MacSyFinder v2: Improved modelling and search engine to identify molecular systems in genomes. bioRxiv, 2022.09.02.506364, ver. 2 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2022.09.02.506364 | MacSyFinder v2: Improved modelling and search engine to identify molecular systems in genomes | Bertrand Néron, Rémi Denise, Charles Coluzzi, Marie Touchon, Eduardo P. C. Rocha, Sophie S. Abby | <p style="text-align: justify;">Complex cellular functions are usually encoded by a set of genes in one or a few organized genetic loci in microbial genomes. Macromolecular System Finder (MacSyFinder) is a program that uses these properties to mod... | | Bacteria and archaea, Bioinformatics, Functional genomics | Gavin Douglas | Kwee Boon Brandon Seah, Max Emil Schön | 2022-09-09 10:30:31 | |
14 Sep 2023
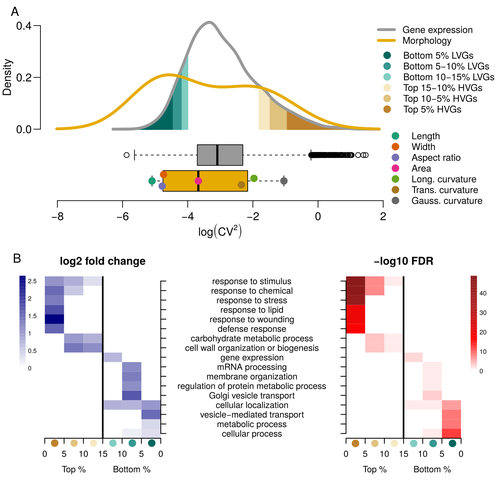
Expression of cell-wall related genes is highly variable and correlates with sepal morphologyThe same but different: How small scale hidden variations can have large effectsRecommended by Francois Sabot based on reviews by Sandra Corjito and 1 anonymous reviewer
For ages, we considered only single genes, or just a few, in order to understand the relationship between phenotype and genotype in response to environmental challenges. Recently, the use of meaningful groups of genes, e.g. gene regulatory networks, or modules of co-expression, allowed scientists to have a larger view of gene regulation. However, all these findings were based on contrasted genotypes, e.g. between wild-types and mutants, as the implicit assumption often made is that there is little transcriptomic variability within the same genotype context. Hartasànchez and collaborators (2023) decided to challenge both views: they used a single genotype instead of two, the famous A. thaliana Col0, and numerous plants, and considered whole gene networks related to sepal morphology and its variations. They used a clever approach, combining high-level phenotyping and gene expression to better understand phenomena and regulations underlying sepal morphologies. Using multiple controls, they showed that basic variations in the expression of genes related to the cell wall regulation, as well as the ones involved in chloroplast metabolism, influenced the global transcriptomic pattern observed in sepal while being in near-identical genetic background and controlling for all other experimental conditions. The paper of Hartasànchez et al. is thus a tremendous call for humility in biology, as we saw in their work that we just understand the gross machinery. However, the Devil is in the details: understanding those very small variations that may have a large influence on phenotypes, and thus on local adaptation to environmental challenges, is of great importance in these times of climatic changes. References Hartasánchez DA, Kiss A, Battu V, Soraru C, Delgado-Vaquera A, Massinon F, Brasó-Vives M, Mollier C, Martin-Magniette M-L, Boudaoud A, Monéger F. 2023. Expression of cell-wall related genes is highly variable and correlates with sepal morphology. bioRxiv, ver. 4, peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2022.04.26.489498 | Expression of cell-wall related genes is highly variable and correlates with sepal morphology | Diego A. Hartasánchez, Annamaria Kiss, Virginie Battu, Charline Soraru, Abigail Delgado-Vaquera, Florian Massinon, Marina Brasó-Vives, Corentin Mollier, Marie-Laure Martin-Magniette, Arezki Boudaoud, Françoise Monéger | <p style="text-align: justify;">Control of organ morphology is a fundamental feature of living organisms. There is, however, observable variation in organ size and shape within a given genotype. Taking the sepal of Arabidopsis as a model, we inves... | | Bioinformatics, Epigenomics, Plants | Francois Sabot | 2023-03-14 19:10:15 |




