
DUPLESSIS Sebastien
- Tree-Microbe Interactions Department - Ecogenomics team, INRAE, Champenoux, France
- Evolutionary genomics, Functional genomics, Fungi, Plants
- recommender
Recommendations: 2
Reviews: 0
Recommendations: 2

Botrytis cinerea strains infecting grapevine and tomato display contrasted repertoires of accessory chromosomes, transposons and small RNAs
Exploring genomic determinants of host specialization in Botrytis cinerea
Recommended by Sebastien Duplessis based on reviews by Cecile Lorrain and Thorsten LangnerThe genomics era has pushed forward our understanding of fungal biology. Much progress has been made in unraveling new gene functions and pathways, as well as the evolution or adaptation of fungi to their hosts or environments through population studies (Hartmann et al. 2019; Gladieux et al. 2018). Closing gaps more systematically in draft genomes using the most recent long-read technologies now seems the new standard, even with fungal species presenting complex genome structures (e.g. large and highly repetitive dikaryotic genomes; Duan et al. 2022). Understanding the genomic dynamics underlying host specialization in phytopathogenic fungi is of utmost importance as it may open new avenues to combat diseases. A strong host specialization is commonly observed for biotrophic and hemi-biotrophic fungal species or for necrotrophic fungi with a narrow host range, whereas necrotrophic fungi with broad host range are considered generalists (Liang and Rollins, 2018; Newman and Derbyshire, 2020). However, some degrees of specialization towards given hosts have been reported in generalist fungi and the underlying mechanisms remain to be determined.
Botrytis cinerea is a polyphagous necrotrophic phytopathogen with a particularly wide host range and it is notably responsible for grey mould disease on many fruits, such as tomato and grapevine. Because of its importance as a plant pathogen, its relatively small genome size and its taxonomical position, it has been targeted for early genome sequencing and a first reference genome was provided in 2011 (Amselem et al. 2011). Other genomes were subsequently sequenced for other strains, and most importantly a gapless assembled version of the initial reference genome B05.10 was provided to the community (van Kan et al. 2017). This genomic resource has supported advances in various aspects of the biology of B. cinerea such as the production of specialized metabolites, which plays an important role in host-plant colonization, or more recently in the production of small RNAs which interfere with the host immune system, representing a new class of non-proteinaceous virulence effectors (Dalmais et al. 2011; Weiberg et al. 2013).
In the present study, Simon et al. (2022) use PacBio long-read sequencing for Sl3 and Vv3 strains, which represent genetic clusters in B. cinerea populations found on tomato and grapevine. The authors combined these complete and high-quality genome assemblies with the B05.10 reference genome and population sequencing data to perform a comparative genomic analysis of specialization towards the two host plants. Transposable elements generate genomic diversity due to their mobile and repetitive nature and they are of utmost importance in the evolution of fungi as they deeply reshape the genomic landscape (Lorrain et al. 2021). Accessory chromosomes are also known drivers of adaptation in fungi (Möller and Stukenbrock, 2017). Here, the authors identify several genomic features such as the presence of different sets of accessory chromosomes, the presence of differentiated repertoires of transposable elements, as well as related small RNAs in the tomato and grapevine populations, all of which may be involved in host specialization. Whereas core chromosomes are highly syntenic between strains, an accessory chromosome validated by pulse-field electrophoresis is specific of the strains isolated from grapevine. Particularly, they show that two particular retrotransposons are discriminant between the strains and that they allow the production of small RNAs that may act as effectors. The discriminant accessory chromosome of the Vv3 strain harbors one of the unraveled retrotransposons as well as new genes of yet unidentified function.
I recommend this article because it perfectly illustrates how efforts put into generating reference genomic sequences of higher quality can lead to new discoveries and allow to build strong hypotheses about biology and evolution in fungi. Also, the study combines an up-to-date genomics approach with a classical methodology such as pulse-field electrophoresis to validate the presence of accessory chromosomes. A major input of this investigation of the genomic determinants of B. cinerea is that it provides solid hints for further analysis of host-specialization at the population level in a broad-scale phytopathogenic fungus.
References
Amselem J, Cuomo CA, Kan JAL van, Viaud M, Benito EP, Couloux A, Coutinho PM, Vries RP de, Dyer PS, Fillinger S, Fournier E, Gout L, Hahn M, Kohn L, Lapalu N, Plummer KM, Pradier J-M, Quévillon E, Sharon A, Simon A, Have A ten, Tudzynski B, Tudzynski P, Wincker P, Andrew M, Anthouard V, Beever RE, Beffa R, Benoit I, Bouzid O, Brault B, Chen Z, Choquer M, Collémare J, Cotton P, Danchin EG, Silva CD, Gautier A, Giraud C, Giraud T, Gonzalez C, Grossetete S, Güldener U, Henrissat B, Howlett BJ, Kodira C, Kretschmer M, Lappartient A, Leroch M, Levis C, Mauceli E, Neuvéglise C, Oeser B, Pearson M, Poulain J, Poussereau N, Quesneville H, Rascle C, Schumacher J, Ségurens B, Sexton A, Silva E, Sirven C, Soanes DM, Talbot NJ, Templeton M, Yandava C, Yarden O, Zeng Q, Rollins JA, Lebrun M-H, Dickman M (2011) Genomic Analysis of the Necrotrophic Fungal Pathogens Sclerotinia sclerotiorum and Botrytis cinerea. PLOS Genetics, 7, e1002230. https://doi.org/10.1371/journal.pgen.1002230
Dalmais B, Schumacher J, Moraga J, Le Pêcheur P, Tudzynski B, Collado IG, Viaud M (2011) The Botrytis cinerea phytotoxin botcinic acid requires two polyketide synthases for production and has a redundant role in virulence with botrydial. Molecular Plant Pathology, 12, 564–579. https://doi.org/10.1111/j.1364-3703.2010.00692.x
Duan H, Jones AW, Hewitt T, Mackenzie A, Hu Y, Sharp A, Lewis D, Mago R, Upadhyaya NM, Rathjen JP, Stone EA, Schwessinger B, Figueroa M, Dodds PN, Periyannan S, Sperschneider J (2022) Physical separation of haplotypes in dikaryons allows benchmarking of phasing accuracy in Nanopore and HiFi assemblies with Hi-C data. Genome Biology, 23, 84. https://doi.org/10.1186/s13059-022-02658-2
Gladieux P, Condon B, Ravel S, Soanes D, Maciel JLN, Nhani A, Chen L, Terauchi R, Lebrun M-H, Tharreau D, Mitchell T, Pedley KF, Valent B, Talbot NJ, Farman M, Fournier E (2018) Gene Flow between Divergent Cereal- and Grass-Specific Lineages of the Rice Blast Fungus Magnaporthe oryzae. mBio, 9, e01219-17. https://doi.org/10.1128/mBio.01219-17
Hartmann FE, Rodríguez de la Vega RC, Carpentier F, Gladieux P, Cornille A, Hood ME, Giraud T (2019) Understanding Adaptation, Coevolution, Host Specialization, and Mating System in Castrating Anther-Smut Fungi by Combining Population and Comparative Genomics. Annual Review of Phytopathology, 57, 431–457. https://doi.org/10.1146/annurev-phyto-082718-095947
Liang X, Rollins JA (2018) Mechanisms of Broad Host Range Necrotrophic Pathogenesis in Sclerotinia sclerotiorum. Phytopathology®, 108, 1128–1140. https://doi.org/10.1094/PHYTO-06-18-0197-RVW
Lorrain C, Oggenfuss U, Croll D, Duplessis S, Stukenbrock E (2021) Transposable Elements in Fungi: Coevolution With the Host Genome Shapes, Genome Architecture, Plasticity and Adaptation. In: Encyclopedia of Mycology (eds Zaragoza Ó, Casadevall A), pp. 142–155. Elsevier, Oxford. https://doi.org/10.1016/B978-0-12-819990-9.00042-1
Möller M, Stukenbrock EH (2017) Evolution and genome architecture in fungal plant pathogens. Nature Reviews Microbiology, 15, 756–771. https://doi.org/10.1038/nrmicro.2017.76
Newman TE, Derbyshire MC (2020) The Evolutionary and Molecular Features of Broad Host-Range Necrotrophy in Plant Pathogenic Fungi. Frontiers in Plant Science, 11. https://doi.org/10.3389/fpls.2020.591733
Simon A, Mercier A, Gladieux P, Poinssot B, Walker A-S, Viaud M (2022) Botrytis cinerea strains infecting grapevine and tomato display contrasted repertoires of accessory chromosomes, transposons and small RNAs. bioRxiv, 2022.03.07.483234, ver. 4 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2022.03.07.483234
Van Kan JAL, Stassen JHM, Mosbach A, Van Der Lee TAJ, Faino L, Farmer AD, Papasotiriou DG, Zhou S, Seidl MF, Cottam E, Edel D, Hahn M, Schwartz DC, Dietrich RA, Widdison S, Scalliet G (2017) A gapless genome sequence of the fungus Botrytis cinerea. Molecular Plant Pathology, 18, 75–89. https://doi.org/10.1111/mpp.12384
Weiberg A, Wang M, Lin F-M, Zhao H, Zhang Z, Kaloshian I, Huang H-D, Jin H (2013) Fungal Small RNAs Suppress Plant Immunity by Hijacking Host RNA Interference Pathways. Science, 342, 118–123. https://doi.org/10.1126/science.1239705
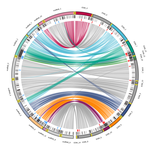
Chromosomal rearrangements with stable repertoires of genes and transposable elements in an invasive forest-pathogenic fungus
Comparative genomics in the chestnut blight fungus Cryphonectria parasitica reveals large chromosomal rearrangements and a stable genome organization
Recommended by Sebastien Duplessis based on reviews by Benjamin Schwessinger and 1 anonymous reviewerAbout twenty-five years after the sequencing of the first fungal genome and a dozen years after the first plant pathogenic fungi genomes were sequenced, unprecedented international efforts have led to an impressive collection of genomes available for the community of mycologists in international databases (Goffeau et al. 1996, Dean et al. 2005; Spatafora et al. 2017). For instance, to date, the Joint Genome Institute Mycocosm database has collected more than 2,100 fungal genomes over the fungal tree of life (https://mycocosm.jgi.doe.gov). Such resources are paving the way for comparative genomics, population genomics and phylogenomics to address a large panel of questions regarding the biology and the ecology of fungal species. Early on, population genomics applied to pathogenic fungi revealed a great diversity of genome content and organization and a wide variety of variants and rearrangements (Raffaele and Kamoun 2012, Hartmann 2022). Such plasticity raises questions about how to choose a representative genome to serve as an ideal reference to address pertinent biological questions.
Cryphonectria parasitica is a fungal pathogen that is infamous for the devastation of chestnut forests in North America after its accidental introduction more than a century ago (Anagnostakis 1987). Since then, it has been a quarantine species under surveillance in various parts of the world. As for other fungi causing diseases on forest trees, the study of adaptation to its host in the forest ecosystem and of its reproduction and dissemination modes is more complex than for crop-targeting pathogens. A first reference genome was published in 2020 for the chestnut blight fungus C. parasitica strain EP155 in the frame of an international project with the DOE JGI (Crouch et al. 2020). Another genome was then sequenced from the French isolate YVO003, which showed a few differences in the assembly suggesting possible rearrangements (Demené et al. 2019). Here the sequencing of a third isolate ESM015 from the native area of C. parasitica in Japan allows to draw broader comparative analysis and particularly to compare between native and introduced isolates (Demené et al. 2022).
Demené and collaborators report on a new genome sequence using up-to-date long-read sequencing technologies and they provide an improved genome assembly. Comparison with previously published C. parasitica genomes did not reveal dramatic changes in the overall chromosomal landscapes, but large rearrangements could be spotted. Despite these rearrangements, the genome content and organization – i.e. genes and repeats – remain stable, with a limited number of genes gains and losses. As in any fungal plant pathogen genome, the repertoire of candidate effectors predicted among secreted proteins was more particularly scrutinized. Such effector genes have previously been reported in other pathogens in repeat-enriched plastic genomic regions with accelerated evolutionary rates under the pressure of the host immune system (Raffaele and Kamoun 2012). Demené and collaborators established a list of priority candidate effectors in the C. parasitica gene catalog likely involved in the interaction with the host plant which will require more attention in future functional studies. Six major inter-chromosomal translocations were detected and are likely associated with double break strands repairs. The authors speculate on the possible effects that these translocations may have on gene organization and expression regulation leading to dramatic phenotypic changes in relation to introduction and invasion in new continents and the impact regarding sexual reproduction in this fungus (Demené et al. 2022).
I recommend this article not only because it is providing an improved assembly of a reference genome for C. parasitica, but also because it adds diversity in terms of genome references availability, with a third high-quality assembly. Such an effort in the tree pathology community for a pathogen under surveillance is of particular importance for future progress in post-genomic analysis, e.g. in further genomic population studies (Hartmann 2022).
References
Anagnostakis SL (1987) Chestnut Blight: The Classical Problem of an Introduced Pathogen. Mycologia, 79, 23–37. https://doi.org/10.2307/3807741
Crouch JA, Dawe A, Aerts A, Barry K, Churchill ACL, Grimwood J, Hillman BI, Milgroom MG, Pangilinan J, Smith M, Salamov A, Schmutz J, Yadav JS, Grigoriev IV, Nuss DL (2020) Genome Sequence of the Chestnut Blight Fungus Cryphonectria parasitica EP155: A Fundamental Resource for an Archetypical Invasive Plant Pathogen. Phytopathology®, 110, 1180–1188. https://doi.org/10.1094/PHYTO-12-19-0478-A
Dean RA, Talbot NJ, Ebbole DJ, Farman ML, Mitchell TK, Orbach MJ, Thon M, Kulkarni R, Xu J-R, Pan H, Read ND, Lee Y-H, Carbone I, Brown D, Oh YY, Donofrio N, Jeong JS, Soanes DM, Djonovic S, Kolomiets E, Rehmeyer C, Li W, Harding M, Kim S, Lebrun M-H, Bohnert H, Coughlan S, Butler J, Calvo S, Ma L-J, Nicol R, Purcell S, Nusbaum C, Galagan JE, Birren BW (2005) The genome sequence of the rice blast fungus Magnaporthe grisea. Nature, 434, 980–986. https://doi.org/10.1038/nature03449
Demené A., Laurent B., Cros-Arteil S., Boury C. and Dutech C. 2022. Chromosomal rearrangements with stable repertoires of genes and transposable elements in an invasive forest-pathogenic fungus. bioRxiv, 2021.03.09.434572, ver.6 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2021.03.09.434572
Goffeau A, Barrell BG, Bussey H, Davis RW, Dujon B, Feldmann H, Galibert F, Hoheisel JD, Jacq C, Johnston M, Louis EJ, Mewes HW, Murakami Y, Philippsen P, Tettelin H, Oliver SG (1996) Life with 6000 Genes. Science, 274, 546–567. https://doi.org/10.1126/science.274.5287.546
Hartmann FE (2022) Using structural variants to understand the ecological and evolutionary dynamics of fungal plant pathogens. New Phytologist, 234, 43–49. https://doi.org/10.1111/nph.17907
Raffaele S, Kamoun S (2012) Genome evolution in filamentous plant pathogens: why bigger can be better. Nature Reviews Microbiology, 10, 417–430. https://doi.org/10.1038/nrmicro2790
Spatafora JW, Aime MC, Grigoriev IV, Martin F, Stajich JE, Blackwell M (2017) The Fungal Tree of Life: from Molecular Systematics to Genome-Scale Phylogenies. Microbiology Spectrum, 5, 5.5.03. https://doi.org/10.1128/microbiolspec.FUNK-0053-2016