
Latest recommendations

| Id | Title▼ | Authors | Abstract | Picture | Thematic fields | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
06 Jul 2021
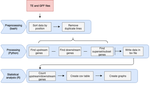
A pipeline to detect the relationship between transposable elements and adjacent genes in host genomesA new tool to cross and analyze TE and gene annotationsRecommended by Emmanuelle Lerat based on reviews by 2 anonymous reviewersTransposable elements (TEs) are important components of genomes. Indeed, they are now recognized as having a major role in gene and genome evolution (Biémont 2010). In particular, several examples have shown that the presence of TEs near genes may influence their functioning, either by recruiting particular epigenetic modifications (Guio et al. 2018) or by directly providing new regulatory sequences allowing new expression patterns (Chung et al. 2007; Sundaram et al. 2014). Therefore, the study of the interaction between TEs and their host genome requires tools to easily cross-annotate both types of entities. In particular, one needs to be able to identify all TEs located in the close vicinity of genes or inside them. Such task may not always be obvious for many biologists, as it requires informatics knowledge to develop their own script codes. In their work, Meguerdichian et al. (2021) propose a command-line pipeline that takes as input the annotations of both genes and TEs for a given genome, then detects and reports the positional relationships between each TE insertion and their closest genes. The results are processed into an R script to provide tables displaying some statistics and graphs to visualize these relationships. This tool has the potential to be very useful for performing preliminary analyses before studying the impact of TEs on gene functioning, especially for biologists. Indeed, it makes it possible to identify genes close to TE insertions. These identified genes could then be specifically considered in order to study in more detail the link between the presence of TEs and their functioning. For example, the identification of TEs close to genes may allow to determine their potential role on gene expression. References Biémont C (2010). A brief history of the status of transposable elements: from junk DNA to major players in evolution. Genetics, 186, 1085–1093. https://doi.org/10.1534/genetics.110.124180 Chung H, Bogwitz MR, McCart C, Andrianopoulos A, ffrench-Constant RH, Batterham P, Daborn PJ (2007). Cis-regulatory elements in the Accord retrotransposon result in tissue-specific expression of the Drosophila melanogaster insecticide resistance gene Cyp6g1. Genetics, 175, 1071–1077. https://doi.org/10.1534/genetics.106.066597 Guio L, Vieira C, González J (2018). Stress affects the epigenetic marks added by natural transposable element insertions in Drosophila melanogaster. Scientific Reports, 8, 12197. https://doi.org/10.1038/s41598-018-30491-w Meguerditchian C, Ergun A, Decroocq V, Lefebvre M, Bui Q-T (2021). A pipeline to detect the relationship between transposable elements and adjacent genes in host genomes. bioRxiv, 2021.02.25.432867, ver. 4 peer-reviewed and recommended by Peer Community In Genomics. https://doi.org/10.1101/2021.02.25.432867 Sundaram V, Cheng Y, Ma Z, Li D, Xing X, Edge P, Snyder MP, Wang T (2014). Widespread contribution of transposable elements to the innovation of gene regulatory networks. Genome Research, 24, 1963–1976. https://doi.org/10.1101/gr.168872.113 | A pipeline to detect the relationship between transposable elements and adjacent genes in host genomes | Caroline Meguerditchian, Ayse Ergun, Veronique Decroocq, Marie Lefebvre, Quynh-Trang Bui | <p>Understanding the relationship between transposable elements (TEs) and their closest positional genes in the host genome is a key point to explore their potential role in genome evolution. Transposable elements can regulate and affect gene expr... | | Bioinformatics, Viruses and transposable elements | Emmanuelle Lerat | 2021-03-03 15:08:34 | ||
23 Aug 2022

A novel lineage of the Capra genus discovered in the Taurus Mountains of Turkey using ancient genomicsGoat ancient DNA analysis unveils a new lineage that may have hybridized with domestic goatsRecommended by Laura Botigué based on reviews by Torsten Günther and 1 anonymous reviewerThe genomic analysis of ancient remains has revolutionized the study of the past over the last decade. On top of the discoveries related to human evolution, plant and animal archaeogenomics has been used to gain new insights into the domestication process and the dispersal of domestic forms. In this study, Daly and colleagues analyse the genomic data from seven goat specimens from the Epipalaeolithic recovered from the Direkli Cave in the Taurus Mountains in southern Turkey. They also generate new genomic data from Capra lineages across the phylogeny, contributing to the availability of genomic resources for this genus. Analysis of the ancient remains is compared to modern genomic variability and sheds light on the complexity of the Tur wild Capra lineages and their relationship with domestic goats and their wild ancestors. Authors find that during the Late Pleistocene in the Taurus Mountains wild goats from the Tur lineage, today restricted to the Caucasus region, were not rare and cohabited with Bezoar, the wild goats that are the ancestors of domestic goats. They identify the Direkli Cave specimens as a lineage separate from the A modified D statistic, Dex, is developed to examine the contribution of the ancient Tur lineage in domestic goats through time and space. Dex measures the relative degree of allele sharing, derived specifically in a selected genome or group of genomes, and may have some utility in genera with complex admixture histories or admixture from ghost lineages. Results confirm that Neolithic European goat had an excess of allele sharing with this ancient Tur lineage, something that is absent in contemporary goats eastwards or in modern goats. Interspecific gene flow is not uncommon among mammals, but the case of Capra has the additional motivation of understanding the origins of the domestic species. This work uncovers an ancient Tur lineage that is different from the modern ones and is additionally found in another geographic area. Furthermore, evidence shows that this ancient lineage exhibits substantial amounts of allele sharing with the wild ancestor of the domestic goat, but also with the Neolithic Eurasian domestic goats, highlighting the complexity of the domestication process. This work has also important implications in understanding the effect of over-hunting and habitat disruption during the Anthropocene on the evolution of the Capra genus. The availability of more ancient specimens and better coverage of the modern genomic variability can help quantifying the lineages that went lost and identify the causes of their extinction. This work is limited by the current availability of whole genomes from modern Capra specimens, but pieces of evidence as well that an effort is needed to obtain more genomic data from ancient goats from different geographic ranges to determine to what extent these lineages contributed to goat domestication. References Daly KG, Arbuckle BS, Rossi C, Mattiangeli V, Lawlor PA, Mashkour M, Sauer E, Lesur J, Atici L, Cevdet CM and Bradley DG (2022) A novel lineage of the Capra genus discovered in the Taurus Mountains of Turkey using ancient genomics. bioRxiv, 2022.04.08.487619, ver. 5 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2022.04.08.487619 | A novel lineage of the Capra genus discovered in the Taurus Mountains of Turkey using ancient genomics | Kevin G. Daly, Benjamin S. Arbuckle, Conor Rossi, Valeria Mattiangeli, Phoebe A. Lawlor, Marjan Mashkour, Eberhard Sauer, Joséphine Lesur, Levent Atici, Cevdet Merih Erek, Daniel G. Bradley | <p>Direkli Cave, located in the Taurus Mountains of southern Turkey, was occupied by Late Epipaleolithic hunters-gatherers for the seasonal hunting and processing of game including large numbers of wild goats. We report genomic data from new and p... | | Evolutionary genomics, Population genomics, Vertebrates | Laura Botigué | 2022-04-15 12:05:47 | ||
09 Oct 2020
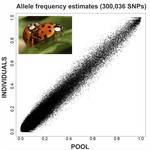
An evaluation of pool-sequencing transcriptome-based exon capture for population genomics in non-model speciesAssessing a novel sequencing-based approach for population genomics in non-model speciesRecommended by Thomas Derrien and Sebastian E. Ramos-Onsins based on reviews by Valentin Wucher and 1 anonymous reviewerDeveloping new sequencing and bioinformatic strategies for non-model species is of great interest in many applications, such as phylogenetic studies of diverse related species, but also for studies in population genomics, where a relatively large number of individuals is necessary. Different approaches have been developed and used in these last two decades, such as RAD-Seq (e.g., Miller et al. 2007), exome sequencing (e.g., Teer and Mullikin 2010) and other genome reduced representation methods that avoid the use of a good reference and well annotated genome (reviewed at Davey et al. 2011). However, population genomics studies require the analysis of numerous individuals, which makes the studies still expensive. Pooling samples was thought as an inexpensive strategy to obtain estimates of variability and other related to the frequency spectrum, thus allowing the study of variability at population level (e.g., Van Tassell et al. 2008), although the major drawback was the loss of information related to the linkage of the variants. In addition, population analysis using all these sequencing strategies require statistical and empirical validations that are not always fully performed. A number of studies aiming to obtain unbiased estimates of variability using reduced representation libraries and/or with pooled data have been performed (e.g., Futschik and Schlötterer 2010, Gautier et al. 2013, Ferretti et al. 2013, Lynch et al. 2014), as well as validation of new sequencing methods for population genetic analyses (e.g., Gautier et al. 2013, Nevado et al. 2014). Nevertheless, empirical validation using both pooled and individual experimental approaches combined with different bioinformatic methods has not been always performed. References [1] Choquet et al. (2019). Towards population genomics in non-model species with large genomes: a case study of the marine zooplankton Calanus finmarchicus. Royal Society open science, 6(2), 180608. doi: https://doi.org/10.1098/rsos.180608 | An evaluation of pool-sequencing transcriptome-based exon capture for population genomics in non-model species | Emeline Deleury, Thomas Guillemaud, Aurélie Blin & Eric Lombaert | <p>Exon capture coupled to high-throughput sequencing constitutes a cost-effective technical solution for addressing specific questions in evolutionary biology by focusing on expressed regions of the genome preferentially targeted by selection. Tr... | | Bioinformatics, Population genomics | Thomas Derrien | 2020-02-26 09:21:11 | ||
06 May 2022

A deep dive into genome assemblies of non-vertebrate animalsDiving, and even digging, into the wild jungle of annotation pathways for non-vertebrate animalsRecommended by Francois Sabot based on reviews by Yann Bourgeois, Cécile Monat, Valentina Peona and Benjamin Istace based on reviews by Yann Bourgeois, Cécile Monat, Valentina Peona and Benjamin Istace
In their paper, Guiglielmoni et al. propose we pick up our snorkels and palms and take "A deep dive into genome assemblies of non-vertebrate animals" (1). Indeed, while numerous assembly-related tools were developed and tested for human genomes (or at least vertebrates such as mice), very few were tested on non-vertebrate animals so far. Moreover, most of the benchmarks are aimed at raw assembly tools, and very few offer a guide from raw reads to an almost finished assembly, including quality control and phasing. This huge and exhaustive review starts with an overview of the current sequencing technologies, followed by the theory of the different approaches for assembly and their implementation. For each approach, the authors present some of the most representative tools, as well as the limits of the approach. The authors additionally present all the steps required to obtain an almost complete assembly at a chromosome-scale, with all the different technologies currently available for scaffolding, QC, and phasing, and the way these tools can be applied to non-vertebrates animals. Finally, they propose some useful advice on the choice of the different approaches (but not always tools, see below), and advocate for a robust genome database with all information on the way the assembly was obtained. This review is a very complete one for now and is a very good starting point for any student or scientist interested to start working on genome assembly, from either model or non-model organisms. However, the authors do not provide a list of tools or a benchmark of them as a recommendation. Why? Because such a proposal may be obsolete in less than a year.... Indeed, with the explosion of the 3rd generation of sequencing technology, assembly tools (from different steps) are constantly evolving, and their relative performance increases on a monthly basis. In addition, some tools are really efficient at the time of a review or of an article, but are not further developed later on, and thus will not evolve with the technology. We have all seen it with wonderful tools such as Chiron (2) or TopHat (3), which were very promising ones, but cannot be developed further due to the stop of the project, the end of the contract of the post-doc in charge of the development, or the decision of the developer to switch to another paradigm. Such advice would, therefore, need to be constantly updated. Thus, the manuscript from Guiglielmoni et al will be an almost intemporal one (up to the next sequencing revolution at last), and as they advocated for a more informed genome database, I think we should consider a rolling benchmarking system (tools, genome and sequence dataset) allowing to keep the performance of the tools up-to-date, and to propose the best set of assembly tools for a given type of genome. References 1. Guiglielmoni N, Rivera-Vicéns R, Koszul R, Flot J-F (2022) A Deep Dive into Genome Assemblies of Non-vertebrate Animals. Preprints, 2021110170, ver. 3 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.20944/preprints202111.0170 2. Teng H, Cao MD, Hall MB, Duarte T, Wang S, Coin LJM (2018) Chiron: translating nanopore raw signal directly into nucleotide sequence using deep learning. GigaScience, 7, giy037. https://doi.org/10.1093/gigascience/giy037 3. Trapnell C, Pachter L, Salzberg SL (2009) TopHat: discovering splice junctions with RNA-Seq. Bioinformatics, 25, 1105–1111. https://doi.org/10.1093/bioinformatics/btp120 | A deep dive into genome assemblies of non-vertebrate animals | Nadège Guiglielmoni, Ramón Rivera-Vicéns, Romain Koszul, Jean-François Flot | <p style="text-align: justify;">Non-vertebrate species represent about ∼95% of known metazoan (animal) diversity. They remain to this day relatively unexplored genetically, but understanding their genome structure and function is pivotal for expan... | | Bioinformatics, Evolutionary genomics | Francois Sabot | Valentina Peona, Benjamin Istace, Cécile Monat, Yann Bourgeois | 2021-11-10 17:47:31 |




