
Latest recommendations

| Id | Title * | Authors * | Abstract * | Picture * | Thematic fields * ▲ | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
09 Aug 2023
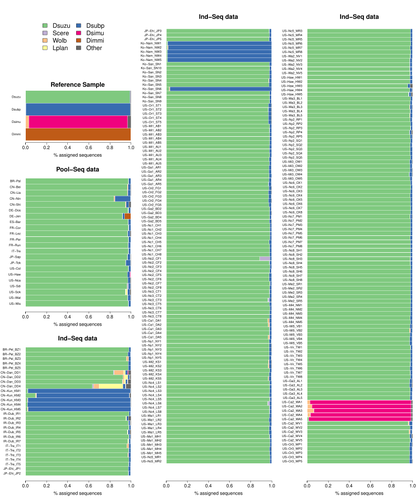
Efficient k-mer based curation of raw sequence data: application in Drosophila suzukiiDecontaminating reads, not contigsRecommended by Nicolas Galtier based on reviews by Marie Cariou and Denis BaurainContamination, the presence of foreign DNA sequences in a sample of interest, is currently a major problem in genomics. Because contamination is often unavoidable at the experimental stage, it is increasingly recognized that the processing of high-throughput sequencing data must include a decontamination step. This is usually performed after the many sequence reads have been assembled into a relatively small number of contigs. Dubious contigs are then discarded based on their composition (e.g. GC-content) or because they are highly similar to a known piece of DNA from a foreign species. Here [1], Mathieu Gautier explores a novel strategy consisting in decontaminating reads, not contigs. Why is this promising? Assembly programs and algorithms are complex, and it is not easy to predict, or monitor, how they handle contaminant reads. Ideally, contaminant reads will be assembled into obvious contaminant contigs. However, there might be more complex situations, such as chimeric contigs with alternating genuine and contaminant segments. Decontaminating at the read level, if possible, should eliminate such unfavorable situations where sequence information from contaminant and target samples are intimately intertwined by an assembler. To achieve this aim, Gautier proposes to use methods initially designed for the analysis of metagenomic data. This is pertinent since the decontamination process involves considering a sample as a mixture of different sources of DNA. The programs used here, CLARK and CLARK-L, are based on so-called k-mer analysis, meaning that the similarity between a read to annotate and a reference sequence is measured by how many sub-sequences (of length 31 base pairs for CLARK and 27 base pairs for CLARK-L) they share. This is notoriously more efficient than traditional sequence alignment algorithms when it comes to comparing a very large number of (most often unrelated) sequences. This is, therefore, a reference-based approach, in which the reads from a sample are assigned to previously sequenced genomes based on k-mer content. This original approach is here specifically applied to the case of Drosophila suzukii, an invasive pest damaging fruit production in Europe and America. Fortunately, Drosophila is a genus of insects with abundant genomic resources, including high-quality reference genomes in dozens of species. Having calibrated and validated his pipeline using data sets of known origins, Gautier quantifies in each of 258 presumed D. suzukii samples the proportion of reads that likely belong to other species of fruit flies, or to fruit fly-associated microbes. This proportion is close to one in 16 samples, which clearly correspond to mis-labelled individuals. It is non-negligible in another ~10 samples, which really correspond to D. suzukii individuals. Most of these reads of unexpected origin are contaminants and should be filtered out. Interestingly, one D. suzukii sample contains a substantial proportion of reads from the closely related D. subpulchera, which might instead reflect a recent episode of gene flow between these two species. The approach, therefore, not only serves as a crucial technical step, but also has the potential to reveal biological processes. Gautier's thorough, well-documented work will clearly benefit the ongoing and future research on D. suzuki, and Drosophila genomics in general. The author and reviewers rightfully note that, like any reference-based approach, this method is heavily dependent on the availability and quality of reference genomes - Drosophila being a favorable case. Building the reference database is a key step, and the interpretation of the output can only be made in the light of its content and gaps, as illustrated by Gautier's careful and detailed discussion of his numerous results. This pioneering study is a striking demonstration of the potential of metagenomic methods for the decontamination of high-throughput sequence data at the read level. The pipeline requires remarkably few computing resources, ensuring low carbon emission. I am looking forward to seeing it applied to a wide range of taxa and samples.
Reference [1] Gautier Mathieu. Efficient k-mer based curation of raw sequence data: application in Drosophila suzukii. bioRxiv, 2023.04.18.537389, ver. 2, peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2023.04.18.537389 | Efficient k-mer based curation of raw sequence data: application in *Drosophila suzukii* | Gautier Mathieu | <p>Several studies have highlighted the presence of contaminated entries in public sequence repositories, calling for special attention to the associated metadata. Here, we propose and evaluate a fast and efficient kmer-based approach to assess th... | | Bioinformatics, Population genomics | Nicolas Galtier | 2023-04-20 22:05:13 | ||
06 Jul 2021
A pipeline to detect the relationship between transposable elements and adjacent genes in host genomesA new tool to cross and analyze TE and gene annotationsRecommended by Emmanuelle Lerat based on reviews by 2 anonymous reviewersTransposable elements (TEs) are important components of genomes. Indeed, they are now recognized as having a major role in gene and genome evolution (Biémont 2010). In particular, several examples have shown that the presence of TEs near genes may influence their functioning, either by recruiting particular epigenetic modifications (Guio et al. 2018) or by directly providing new regulatory sequences allowing new expression patterns (Chung et al. 2007; Sundaram et al. 2014). Therefore, the study of the interaction between TEs and their host genome requires tools to easily cross-annotate both types of entities. In particular, one needs to be able to identify all TEs located in the close vicinity of genes or inside them. Such task may not always be obvious for many biologists, as it requires informatics knowledge to develop their own script codes. In their work, Meguerdichian et al. (2021) propose a command-line pipeline that takes as input the annotations of both genes and TEs for a given genome, then detects and reports the positional relationships between each TE insertion and their closest genes. The results are processed into an R script to provide tables displaying some statistics and graphs to visualize these relationships. This tool has the potential to be very useful for performing preliminary analyses before studying the impact of TEs on gene functioning, especially for biologists. Indeed, it makes it possible to identify genes close to TE insertions. These identified genes could then be specifically considered in order to study in more detail the link between the presence of TEs and their functioning. For example, the identification of TEs close to genes may allow to determine their potential role on gene expression. References Biémont C (2010). A brief history of the status of transposable elements: from junk DNA to major players in evolution. Genetics, 186, 1085–1093. https://doi.org/10.1534/genetics.110.124180 Chung H, Bogwitz MR, McCart C, Andrianopoulos A, ffrench-Constant RH, Batterham P, Daborn PJ (2007). Cis-regulatory elements in the Accord retrotransposon result in tissue-specific expression of the Drosophila melanogaster insecticide resistance gene Cyp6g1. Genetics, 175, 1071–1077. https://doi.org/10.1534/genetics.106.066597 Guio L, Vieira C, González J (2018). Stress affects the epigenetic marks added by natural transposable element insertions in Drosophila melanogaster. Scientific Reports, 8, 12197. https://doi.org/10.1038/s41598-018-30491-w Meguerditchian C, Ergun A, Decroocq V, Lefebvre M, Bui Q-T (2021). A pipeline to detect the relationship between transposable elements and adjacent genes in host genomes. bioRxiv, 2021.02.25.432867, ver. 4 peer-reviewed and recommended by Peer Community In Genomics. https://doi.org/10.1101/2021.02.25.432867 Sundaram V, Cheng Y, Ma Z, Li D, Xing X, Edge P, Snyder MP, Wang T (2014). Widespread contribution of transposable elements to the innovation of gene regulatory networks. Genome Research, 24, 1963–1976. https://doi.org/10.1101/gr.168872.113 | A pipeline to detect the relationship between transposable elements and adjacent genes in host genomes | Caroline Meguerditchian, Ayse Ergun, Veronique Decroocq, Marie Lefebvre, Quynh-Trang Bui | <p>Understanding the relationship between transposable elements (TEs) and their closest positional genes in the host genome is a key point to explore their potential role in genome evolution. Transposable elements can regulate and affect gene expr... | | Bioinformatics, Viruses and transposable elements | Emmanuelle Lerat | 2021-03-03 15:08:34 | ||
13 Jul 2022
Nucleosome patterns in four plant pathogenic fungi with contrasted genome structuresGenome-wide chromatin and expression datasets of various pathogenic ascomycetesRecommended by Sébastien Bloyer and Romain Koszul based on reviews by Ricardo C. Rodríguez de la Vega and 1 anonymous reviewerPlant pathogenic fungi represent serious economic threats. These organisms are rapidly adaptable, with plastic genomes containing many variable regions and evolving rapidly. It is, therefore, useful to characterize their genetic regulation in order to improve their control. One of the steps to do this is to obtain omics data that link their DNA structure and gene expression. Clairet C, Lapalu N, Simon A, Soyer JL, Viaud M, Zehraoui E, Dalmais B, Fudal I, Ponts N (2022) Nucleosome patterns in four plant pathogenic fungi with contrasted genome structures. bioRxiv, 2021.04.16.439968, ver. 4 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2021.04.16.439968 | Nucleosome patterns in four plant pathogenic fungi with contrasted genome structures | Colin Clairet, Nicolas Lapalu, Adeline Simon, Jessica L. Soyer, Muriel Viaud, Enric Zehraoui, Berengere Dalmais, Isabelle Fudal, Nadia Ponts | <p style="text-align: justify;">Fungal pathogens represent a serious threat towards agriculture, health, and environment. Control of fungal diseases on crops necessitates a global understanding of fungal pathogenicity determinants and their expres... | | Epigenomics, Fungi | Sébastien Bloyer | 2021-04-17 10:32:41 | ||
01 Jul 2024
Contextualising samples: Supporting reference genomes of European biodiversity through sample and associated metadata collectionTo avoid biases and to be FAIR, we need to CARE and share biodiversity metadataRecommended by Francois Sabot based on reviews by Julian Osuji and 1 anonymous reviewer based on reviews by Julian Osuji and 1 anonymous reviewer
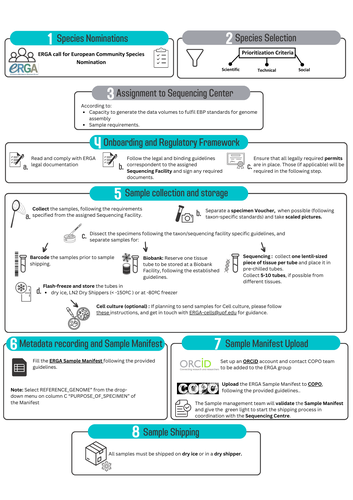
Böhne et al. (2024) do not present a classical scientific paper per se but a report on how the European Reference Genome Atlas (ERGA) aims to deal with sampling and sample information, i.e. metadata. As the goal of ERGA is to provide an almost fully representative set of reference genomes representative of European biodiversity to serve many research areas in biology, they have to be really exhaustive. In this regard, in addition to providing sample metadata recording guidelines, they also discuss the biases existing in sampling and sequencing projects. The first task for such a project is to be sure that the data they generate will be usable and available in the future (“[in] perpetuity", Böhne et al. 2024). The authors deployed a very efficient pipeline for conserving information on sampling: location, physical information, copies of tissues and of DNA, shipping, legal/ethical aspects regarding the Nagoya Protocol, etc., alongside a best-practice manual. This effort is linked to practical guides for the DNA extraction of specific taxa. More generally, these details enable “Findable, Accessible, Interoperable, and Reusable” (FAIR) principles (Wilkinson et al. 2016) to be followed. An important aspect of this paper, in addition to practical points, is the reflection upon the different biases inherent to the choice of sequenced samples. Acknowledging their own biases with regards to DNA extraction protocol efficiency, small genome size choice, as well as the availability of material (Nagoya Protocol aspects) and material transfer efficiency, the authors recommend in the future to not survey biodiversity by selecting one’s favorite samples or species, but also considering "orphan" taxa. Some of these "orphan" taxonomic groups belong to non-arthropod invertebrates but internal disparities are also prominent within other taxa. Finally, the implementation of the "Collective benefit, Authority to control, Responsibility, and Ethics" (CARE) principles (Carroll et al. 2021) will allow Indigenous rights to be considered when prioritizing samples, and to enable their "knowledge systems to permeate throughout the process of reference genome production and beyond" (Böhne et al. 2024). Last, but not least, as ERGA, including its Sampling and Sample Processing committee, is a large collective effort, it is very refreshing to read a paper starting with the acknowledgements and the roles of each member.
References Böhne A, Fernández R, Leonard JA, McCartney AM, McTaggart S, Melo-Ferreira J, Monteiro R, Oomen RA, Pettersson OV, Struck TH (2024) Contextualising samples: Supporting reference genomes of European biodiversity through sample and associated metadata collection. bioRxiv, ver. 3 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2023.06.28.546652 Carroll SR, Herczog E, Hudson M, Russell K, Stall S (2021) Operationalizing the CARE and FAIR Principles for Indigenous data futures. Scientific Data, 8, 108. https://doi.org/10.1038/s41597-021-00892-0 Wilkinson MD, Dumontier M, Aalbersberg IjJ, Appleton G, Axton M, Baak A, Blomberg N, Boiten J-W, da Silva Santos LB, Bourne PE, Bouwman J, Brookes AJ, Clark T, Crosas M, Dillo I, Dumon O, Edmunds S, Evelo CT, Finkers R, Gonzalez-Beltran A, Gray AJG, Groth P, Goble C, Grethe JS, Heringa J, ’t Hoen PAC, Hooft R, Kuhn T, Kok R, Kok J, Lusher SJ, Martone ME, Mons A, Packer AL, Persson B, Rocca-Serra P, Roos M, van Schaik R, Sansone S-A, Schultes E, Sengstag T, Slater T, Strawn G, Swertz MA, Thompson M, van der Lei J, van Mulligen E, Velterop J, Waagmeester A, Wittenburg P, Wolstencroft K, Zhao J, Mons B (2016) The FAIR Guiding Principles for scientific data management and stewardship. Scientific Data, 3, 160018. https://doi.org/10.1038/sdata.2016.18 | Contextualising samples: Supporting reference genomes of European biodiversity through sample and associated metadata collection | Astrid Böhne, Rosa Fernández, Jennifer A. Leonard, Ann M. McCartney, Seanna McTaggart, José Melo-Ferreira, Rita Monteiro, Rebekah A. Oomen, Olga Vinnere Pettersson, Torsten H. Struck | <p>The European Reference Genome Atlas (ERGA) consortium aims to generate a reference genome catalogue for all of Europe's eukaryotic biodiversity. The biological material underlying this mission, the specimens and their derived samples, are provi... | | ERGA, ERGA BGE, ERGA Pilot, Evolutionary genomics | Francois Sabot | Julian Osuji, Francois Sabot, Anonymous | 2023-07-03 10:39:36 | |
20 Nov 2023
Building a Portuguese Coalition for Biodiversity GenomicsThe Portuguese genomics community teams up with iconic species to understand the destruction of biodiversityRecommended by Fernando Racimo based on reviews by Svein-Ole Mikalsen and 1 anonymous reviewerThis manuscript describes the ongoing work and plans of Biogenome Portugal: a new network of researchers in the Portuguese biodiversity genomics community. The aims of this network are to jointly train scientists in ecology and evolution, generate new knowledge and understanding of Portuguese biodiversity, and better engage with the public and with international researchers, so as to advance conservation efforts in the region. In collaboration across disciplines and institutions, they are also contributing to the European Reference Genome Atlas (ERGA): a massive scientific effort, seeking to eventually produce reference-quality genomes for all species in the European continent (Mc Cartney et al. 2023). The manuscript centers around six iconic and/or severely threatened species, whose range extends across parts of what is today considered Portuguese territory. Via the Portugal chapter of ERGA (ERGA-Portugal), the researchers will generate high-quality genome sequences from these species. The species are the Iberian hare, the Azores laurel, the Black wheatear, the Portuguese crowberry, the Cave ground beetle and the Iberian minnowcarp. In ignorance of human-made political borders, some of these species also occupy large parts of the rest of the Iberian peninsula, highlighting the importance of transnational collaboration in biodiversity efforts. The researchers extracted samples from members of each of these species, and are building reference genome sequences from them. In some cases, these sequences will also be co-analyzed with additional population genomic data from the same species or genetic data from cohabiting species. The researchers aim to answer a variety of ecological and evolutionary questions using this information, including how genetic diversity is being affected by the destruction of their habitat, and how they are being forced to adapt as a consequence of the climate emergency. The authors did a very good job in providing a justification for the choice of pilot species, a thorough methodological overview of current work, and well thought-out plans for future analyses once the genome sequences are available for study. The authors also describe plans for networking and training activities to foster a well-connected Portuguese biodiversity genomics community. Applying a genomic analysis lens is important for understanding the ever faster process of devastation of our natural world. Governments and corporations around the globe are destroying nature at ever larger scales (Diaz et al. 2019). They are also destabilizing the climatic conditions on which life has existed for thousands of years (Trisos et al. 2020). Thus, genetic diversity is decreasing faster than ever in human history, even when it comes to non-threatened species (Exposito-Alonso et al. 2022), and these decreases are disrupting ecological processes worldwide (Richardson et al. 2023). This, in turn, is threatening the conditions on which the stability of our societies rest (Gardner and Bullock 2021). The efforts of Biogenome Portal and ERGA-Portugal will go a long way in helping us understand in greater detail how this process is unfolding in Portuguese territories.
References Díaz, Sandra, et al. "Pervasive human-driven decline of life on Earth points to the need for transformative change." Science 366.6471 (2019): eaax3100. https://doi.org/10.1126/science.aax3100 Exposito-Alonso, Moises, et al. "Genetic diversity loss in the Anthropocene." Science 377.6613 (2022): 1431-1435. https://doi.org/10.1126/science.abn5642 Gardner, Charlie J., and James M. Bullock. "In the climate emergency, conservation must become survival ecology." Frontiers in Conservation Science 2 (2021): 659912. https://doi.org/10.3389/fcosc.2021.659912 Mc Cartney, Ann M., et al. "The European Reference Genome Atlas: piloting a decentralised approach to equitable biodiversity genomics." bioRxiv (2023): 2023-09, ver. 2 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.32942/X20W3Q Richardson, Katherine, et al. "Earth beyond six of nine planetary boundaries." Science Advances 9.37 (2023): eadh2458. https://doi.org/10.1126/sciadv.adh2458 Trisos, Christopher H., Cory Merow, and Alex L. Pigot. "The projected timing of abrupt ecological disruption from climate change." Nature 580.7804 (2020): 496-501. https://doi.org/10.1038/s41586-020-2189-9 | Building a Portuguese Coalition for Biodiversity Genomics | João Pedro Marques, Paulo Célio Alves, Isabel R. Amorim, Ricardo J. Lopes, Mónica Moura, Gene Meyers, Manuela Sim-Sim, Carla Sousa-Santos, Maria Judite Alves, Paulo AV Borges, Thomas Brown, Miguel Carneiro, Carlos Carrapato, Luís Ceríaco, Claudio ... | <p style="text-align: justify;">The diverse physiography of the Portuguese land and marine territory, spanning from continental Europe to the Atlantic archipelagos, has made it an important repository of biodiversity throughout the Pleistocene gla... | | ERGA, ERGA Pilot | Fernando Racimo | 2023-07-14 11:24:22 | ||
06 Feb 2024

The need of decoding life for taking care of biodiversity and the sustainable use of nature in the Anthropocene - a Faroese perspectiveWhy sequence everything? A raison d’être for the Genome Atlas of Faroese EcologyRecommended by Stephen Richards based on reviews by Tereza Manousaki and 1 anonymous reviewer
When discussing the Earth BioGenome Project with scientists and potential funding agencies, one common question is: why sequence everything? Whether sequencing a subset would be more optimal is not an unreasonable question given what we know about the mathematics of importance and Pareto’s 80:20 principle, that 80% of the benefits can come from 20% of the effort. However, one must remember that this principle is an observation made in hindsight and selecting the most effective 20% of experiments is difficult. As an example, few saw great applied value in comparative genomic analysis of the archaea Haloferax mediterranei, but this enabled the discovery of CRISPR/Cas9 technology (1). When discussing whether or not to sequence all life on our planet, smaller countries such as the Faroe Islands are seldom mentioned.
1 Mojica, F. J., Díez-Villaseñor, C. S., García-Martínez, J. & Soria, E. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J Mol Evol 60, 174-182 (2005). 2 Mikalsen, S-O., Hjøllum, J. í., Salter, I., Djurhuus, A. & Kongsstovu, S. í. The need of decoding life for taking care of biodiversity and the sustainable use of nature in the Anthropocene – a Faroese perspective. EcoEvoRxiv (2024), ver. 3 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.32942/X21S4C | The need of decoding life for taking care of biodiversity and the sustainable use of nature in the Anthropocene - a Faroese perspective | Svein-Ole Mikalsen, Jari í Hjøllum, Ian Salter, Anni Djurhuus, Sunnvør í Kongsstovu | <p>Biodiversity is under pressure, mainly due to human activities and climate change. At the international policy level, it is now recognised that genetic diversity is an important part of biodiversity. The availability of high-quality reference g... | | ERGA, ERGA Pilot, Population genomics, Vertebrates | Stephen Richards | 2023-07-31 16:59:33 | ||
15 Jan 2024
The genome sequence of the Montseny horsehair worm, Gordionus montsenyensis sp. nov., a key resource to investigate Ecdysozoa evolutionEmbarking on a novel journey in Metazoa evolution through the pioneering sequencing of a key underrepresented lineageRecommended by Juan C. Opazo based on reviews by Gonzalo Riadi and 2 anonymous reviewers
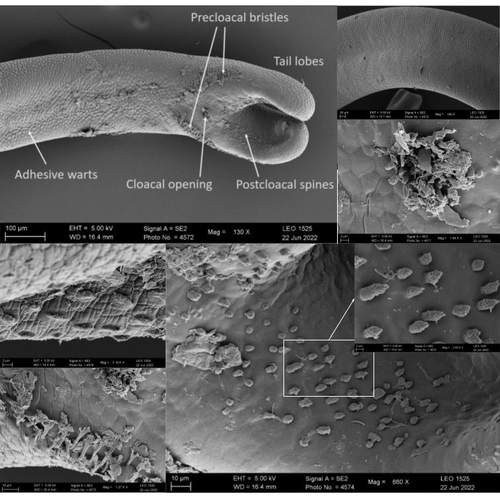
Whole genome sequences are revolutionizing our understanding across various biological fields. They not only shed light on the evolution of genetic material but also uncover the genetic basis of phenotypic diversity. The sequencing of underrepresented lineages, such as the one presented in this study, is of critical importance. It is crucial in filling significant gaps in our understanding of Metazoa evolution. Despite the wealth of genome sequences in public databases, it is crucial to acknowledge that some lineages across the Tree of Life are underrepresented or absent. This research represents a significant step towards addressing this imbalance, contributing to the collective knowledge of the global scientific community. In this genome note, as part of the European Reference Genome Atlas pilot effort to generate reference genomes for European biodiversity (Mc Cartney et al. 2023), Klara Eleftheriadi and colleagues (Eleftheriadi et al. 2023) make a significant effort to add a genome sequence of an unrepresented group in the animal Tree of Life. More specifically, they present a taxonomic description and chromosome-level genome assembly of a newly described species of horsehair worm (Gordionus montsenyensis). Their sequence methodology gave rise to an assembly of 396 scaffolds totaling 288 Mb, with an N50 value of 64.4 Mb, where 97% of this assembly is grouped into five pseudochromosomes. The nuclear genome annotation predicted 10,320 protein-coding genes, and they also assembled the circular mitochondrial genome into a 15-kilobase sequence. The selection of a species representing the phylum Nematomorpha, a group of parasitic organisms belonging to the Ecdysozoa lineage, is good, since today, there is only one publicly available genome for this animal phylum (Cunha et al. 2023). Interestingly, this article shows, among other things, that the species analyzed has lost ∼30% of the universal Metazoan genes. Efforts, like the one performed by Eleftheriadi and colleagues, are necessary to gain more insights, for example, on the evolution of this massive gene lost in this group of animals.
Cunha, T. J., de Medeiros, B. A. S, Lord, A., Sørensen, M. V., and Giribet, G. (2023). Rampant Loss of Universal Metazoan Genes Revealed by a Chromosome-Level Genome Assembly of the Parasitic Nematomorpha. Current Biology, 33 (16): 3514–21.e4. https://doi.org/10.1016/j.cub.2023.07.003 Eleftheriadi, K., Guiglielmoni, N., Salces-Ortiz, J., Vargas-Chavez, C., Martínez-Redondo, G. I., Gut, M., Flot, J.-F., Schmidt-Rhaesa, A., and Fernández, R. (2023). The Genome Sequence of the Montseny Horsehair worm, Gordionus montsenyensis sp. Nov., a Key Resource to Investigate Ecdysozoa Evolution. bioRxiv, ver. 3 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2023.06.26.546503 Mc Cartney, A. M., Formenti, G., Mouton, A., De Panis, D., Marins, L. S., Leitão, H. G., Diedericks, G., et al. (2023). The European Reference Genome Atlas: Piloting a Decentralised Approach to Equitable Biodiversity Genomics. bioRxiv. https://doi.org/10.1101/2023.09.25.559365 | The genome sequence of the Montseny horsehair worm, *Gordionus montsenyensis* sp. nov., a key resource to investigate Ecdysozoa evolution | Eleftheriadi Klara, Guiglielmoni Nadège, Salces-Ortiz Judit, Vargas-Chávez Carlos, Martínez-Redondo Gemma I, Gut Marta, Flot Jean François, Schmidt-Rhaesa Andreas, Fernández Rosa | <p>Nematomorpha, also known as Gordiacea or Gordian worms, are a phylum of parasitic organisms that belong to the Ecdysozoa, a clade of invertebrate animals characterized by molting. They are one of the less scientifically studied animal phyla, an... | | ERGA Pilot | Juan C. Opazo | 2023-06-29 10:31:36 | ||
13 Jul 2024
High quality genome assembly and annotation (v1) of the eukaryotic terrestrial microalga Coccomyxa viridis SAG 216-4Reference genome for the lichen-forming green alga Coccomyxa viridis SAG 216–4Recommended by Iker Irisarri based on reviews by Elisa Goldbecker, Fabian Haas and 2 anonymous reviewers
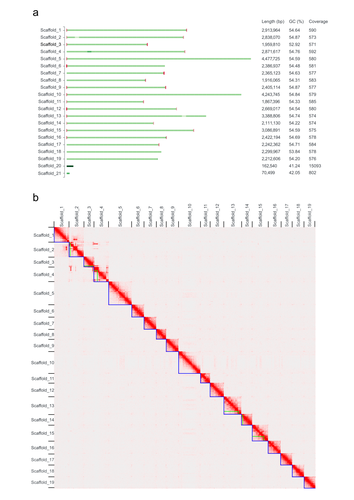
Green algae of the genus Coccomyxa (family Trebouxiophyceae) are extremely diverse in their morphology, habitat (i.e., in marine, freshwater, and terrestrial environments) and lifestyle, including free-living and mutualistic forms. Coccomyxa viridis (strain SAG 216–4) is a photobiont in the lichen Peltigera aphthosa, which was isolated in Switzerland more than 70 years ago (cf. SAG, the Culture Collection of Algae at the University of Göttingen, Germany). Despite the high diversity and plasticity in Coccomyxa, integrative taxonomic analyses led Darienko et al. (2015) to propose clear species boundaries. These authors also showed that symbiotic strains that form lichens evolved multiple times independently in Coccomyxa. Using state-of-the-art sequencing data and bioinformatic methods, including Pac-Bio HiFi and ONT long reads, as well as Hi-C chromatin conformation information, Kraege et al. (2024) generated a high-quality genome assembly for the Coccomyxa viridis strain SAG 216–4. They reconstructed 19 complete nuclear chromosomes, flanked by telomeric regions, totaling 50.9 Mb, plus the plastid and mitochondrial genomes. The performed quality controls leave no doubt of the high quality of the genome assemblies and structural annotations. An interesting observation is the lack of conserved synteny with the close relative Coccomyxa subellipsoidea, but further comparative studies with additional Coccomyxa strains will be required to grasp the genomic evolution in this genus of green algae. This project is framed within the ERGA pilot project, which aims to establish a pan-European genomics infrastructure and contribute to cataloging genomic biodiversity and producing resources that can inform conservation strategies (Formenti et al. 2022). This complete reference genome represents an important step towards this goal, in addition to contributing to future genomic analyses of Coccomyxa more generally.
References Darienko T, Gustavs L, Eggert A, Wolf W, Pröschold T (2015) Evaluating the species boundaries of green microalgae (Coccomyxa, Trebouxiophyceae, Chlorophyta) using integrative taxonomy and DNA barcoding with further implications for the species identification in environmental samples. PLOS ONE, 10, e0127838. https://doi.org/10.1371/journal.pone.0127838 Formenti G, Theissinger K, Fernandes C, Bista I, Bombarely A, Bleidorn C, Ciofi C, Crottini A, Godoy JA, Höglund J, Malukiewicz J, Mouton A, Oomen RA, Paez S, Palsbøll PJ, Pampoulie C, Ruiz-López MJ, Svardal H, Theofanopoulou C, de Vries J, Waldvogel A-M, Zhang G, Mazzoni CJ, Jarvis ED, Bálint M, European Reference Genome Atlas Consortium (2022) The era of reference genomes in conservation genomics. Trends in Ecology & Evolution, 37, 197–202. https://doi.org/10.1016/j.tree.2021.11.008 Kraege A, Chavarro-Carrero EA, Guiglielmoni N, Schnell E, Kirangwa J, Heilmann-Heimbach S, Becker K, Köhrer K, WGGC Team, DeRGA Community, Schiffer P, Thomma BPHJ, Rovenich H (2024) High quality genome assembly and annotation (v1) of the eukaryotic terrestrial microalga Coccomyxa viridis SAG 216-4. bioRxiv, ver. 2 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2023.07.11.548521 | High quality genome assembly and annotation (v1) of the eukaryotic terrestrial microalga *Coccomyxa viridis* SAG 216-4 | Anton Kraege, Edgar Chavarro-Carrero, Nadège Guiglielmoni, Eva Schnell, Joseph Kirangwa, Stefanie Heilmann-Heimbach, Kerstin Becker, Karl Köhrer, Philipp Schiffer, Bart P. H. J. Thomma, Hanna Rovenich | <p>Unicellular green algae of the genus Coccomyxa are recognized for their worldwide distribution and ecological versatility. Most species described to date live in close association with various host species, such as in lichen associations. Howev... | | ERGA Pilot | Iker Irisarri | 2023-11-09 11:54:43 | ||
24 Jan 2024

High quality genome assembly of the brown hare (Lepus europaeus) with chromosome-level scaffoldingA high quality reference genome of the brown hareRecommended by Ed Hollox based on reviews by Merce Montoliu-Nerin and 1 anonymous reviewer
The brown hare, or European hare, Lupus europaeus, is a widespread mammal whose natural range spans western Eurasia. At the northern limit of its range, it hybridises with the mountain hare (L. timidis), and humans have introduced it into other continents. It represents a particularly interesting mammal to study for its population genetics, extensive hybridisation zones, and as an invasive species. This study (Michell et al. 2024) has generated a high-quality assembly of a genome from a brown hare from Finland using long PacBio HiFi sequencing reads and Hi-C scaffolding. The contig N50 of this new genome is 43 Mb, and completeness, assessed using BUSCO, is 96.1%. The assembly comprises 23 autosomes, and an X chromosome and Y chromosome, with many chromosomes including telomeric repeats, indicating the high level of completeness of this assembly. While the genome of the mountain hare has previously been assembled, its assembly was based on a short-read shotgun assembly, with the rabbit as a reference genome. The new high-quality brown hare genome assembly allows a direct comparison with the rabbit genome assembly. For example, the assembly addresses the karyotype difference between the hare (n=24) and the rabbit (n=22). Chromosomes 12 and 17 of the hare are equivalent to chromosome 1 of the rabbit, and chromosomes 13 and 16 of the hare are equivalent to chromosome 2 of the rabbit. The new assembly also provides a hare Y-chromosome, as the previous mountain hare genome was from a female. This new genome assembly provides an important foundation for population genetics and evolutionary studies of lagomorphs. References Michell, C., Collins, J., Laine, P. K., Fekete, Z., Tapanainen, R., Wood, J. M. D., Goffart, S., Pohjoismäki, J. L. O. (2024). High quality genome assembly of the brown hare (Lepus europaeus) with chromosome-level scaffolding. bioRxiv, ver. 3 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2023.08.29.555262 | High quality genome assembly of the brown hare (*Lepus europaeus*) with chromosome-level scaffolding | Craig Michell, Joanna Collins, Pia K. Laine, Zsofia Fekete, Riikka Tapanainen, Jonathan M. D. Wood, Steffi Goffart, Jaakko L. O. Pohjoismaki | <p style="text-align: justify;">We present here a high-quality genome assembly of the brown hare (Lepus europaeus Pallas), based on a fibroblast cell line of a male specimen from Liperi, Eastern Finland. This brown hare genome represents the first... | | ERGA Pilot, Vertebrates | Ed Hollox | 2023-10-16 20:46:39 | ||
06 Apr 2021
Evidence for shared ancestry between Actinobacteria and Firmicutes bacteriophagesViruses of bacteria: phages evolution across phylum boundariesRecommended by Denis Tagu based on reviews by 3 anonymous reviewersBacteria and phages have coexisted and coevolved for a long time. Phages are bacteria-infecting viruses, with a symbiotic status sensu lato, meaning they can be pathogenic, commensal or mutualistic. Thus, the association between bacteria phages has probably played a key role in the high adaptability of bacteria to most - if not all – of Earth’s ecosystems, including other living organisms (such as eukaryotes), and also regulate bacterial community size (for instance during bacterial blooms). As genetic entities, phages are submitted to mutations and natural selection, which changes their DNA sequence. Therefore, comparative genomic analyses of contemporary phages can be useful to understand their evolutionary dynamics. International initiatives such as SEA-PHAGES have started to tackle the issue of history of phage-bacteria interactions and to describe the dynamics of the co-evolution between bacterial hosts and their associated viruses. Indeed, the understanding of this cross-talk has many potential implications in terms of health and agriculture, among others. The work of Koert et al. (2021) deals with one of the largest groups of bacteria (Actinobacteria), which are Gram-positive bacteria mainly found in soil and water. Some soil-born Actinobacteria develop filamentous structures reminiscent of the mycelium of eukaryotic fungi. In this study, the authors focused on the Streptomyces clade, a large genus of Actinobacteria colonized by phages known for their high level of genetic diversity. The authors tested the hypothesis that large exchanges of genetic material occurred between Streptomyces and diverse phages associated with bacterial hosts. Using public datasets, their comparative phylogenomic analyses identified a new cluster among Actinobacteria–infecting phages closely related to phages of Firmicutes. Moreover, the GC content and codon-usage biases of this group of phages of Actinobacteria are similar to those of Firmicutes. This work demonstrates for the first time the transfer of a bacteriophage lineage from one bacterial phylum to another one. The results presented here suggest that the age of the described transfer is probably recent since several genomic characteristics of the phage are not fully adapted to their new hosts. However, the frequency of such transfer events remains an open question. If frequent, such exchanges would mean that pools of bacteriophages are regularly fueled by genetic material coming from external sources, which would have important implications for the co-evolutionary dynamics of phages and bacteria. References Koert, M., López-Pérez, J., Courtney Mattson, C., Caruso, S. and Erill, I. (2021) Evidence for shared ancestry between Actinobacteria and Firmicutes bacteriophages. bioRxiv, 842583, version 5 peer-reviewed and recommended by Peer community in Genomics. doi: https://doi.org/10.1101/842583 | Evidence for shared ancestry between Actinobacteria and Firmicutes bacteriophages | Matthew Koert, Júlia López-Pérez, Courtney Mattson, Steven M. Caruso, Ivan Erill | <p>Bacteriophages typically infect a small set of related bacterial strains. The transfer of bacteriophages between more distant clades of bacteria has often been postulated, but remains mostly unaddressed. In this work we leverage the sequencing ... | | Evolutionary genomics | Denis Tagu | 2019-12-10 15:26:31 |




