
YU Tianxiong 
- Genomics and Computational Biology, University of Massachusetts Chan Medical School, Wocester, United States
- Bioinformatics, Epigenomics, Evolutionary genomics, Population genomics, Viruses and transposable elements
Recommendations: 0
Review: 1
Review: 1
Particular sequence characteristics induce bias in the detection of polymorphic transposable element insertions
A new simulation pipeline enhances benchmarking of transposon polymorphism detection tools
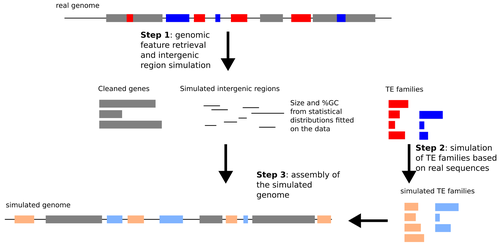
Recommended by Raúl Castanera based on reviews by Tianxiong Yu and 1 anonymous reviewerTransposable Elements (TEs) are one of the main sources of genome variability. However, their study in populations has been hampered by the difficulty of properly detecting them using whole-genome re-sequencing data. Despite the expectations generated by the rise of long-read sequencing, today it is becoming clear that such technologies will not replace short-reads for analyzing large populations in the short term. Detecting Transposon Insertion Polymorphisms (TIPs) from short-read data is a challenging task, due to the repetitive nature of TE sequences that complicate read mapping. Nevertheless, accurate TIP detection is essential for understanding the evolutionary dynamics of TEs, their regulatory roles and their link with phenotypic variability. In the past 15 years, more than 20 tools have been developed for TIP detection using short-read data, but only a few independent benchmarks have been performed so far (Chen et al. 2023; Nelson et al. 2017; Rishishwar et al. 2017; Vendrell-Mir et al. 2019). Previous benchmarks have used simulated and real data to evaluate tool performance, each with its own set of advantages and limitations. In particular, introducing artificial insertions and simulating genomic short-reads may not reflect the nature of real TEs. By contrast, using real TE insertions as benchmarks can introduce bias since TE annotations are never perfect.
Verneret et al. (2025) introduce an original, alternative approach in which a comprehensive simulation method mimics the most important sequence features of real TEs and non-TE intergenic regions. This simulated data is then combined with true genic sequences, generating a pseudochromosome that can be used for benchmarking TIP detection pipelines. Using this approach, the authors eliminate the bias of TE annotation on real genomes, while preserving most of the characteristics of natural TEs. Using simulated pseudochromosomes for Drosophila melanogaster and Arabidopsis thaliana, Verneret et al. (2025) found that the performance of 14 commonly used TIP-calling tools is highly variable, with only a few performing well, and only at high sequencing depths. In addition to this, the authors analyzed the sequence features of true-positive and false-positive TIP calls, and found that specific TE sequence characteristics (e.g., length, age, etc.) affect the detection of both reference and non-reference TIPs.
The approach described by Verneret et al. (2025) is an important contribution to the field for several reasons. On the one hand, the results shown in the publication will help the users of such tools make informed decisions before launching their experiments. For more advanced users, it will enable future benchmarks to identify which tools perform best for different species, each with their own sequence characteristics. For software developers, the data released constitutes a precious dataset to test their tools in the same conditions. Finally, the identification of sequence characteristics enriched among false positives and false negatives also gives an opportunity for developers to improve the performance of the new tools by considering these specificities.
References
Chen J, Basting PJ, Han S, Garfinkel DJ, Bergman CM (2023) Reproducible evaluation of transposable element detectors with McClintock 2 guides accurate inference of Ty insertion patterns in yeast. Mobile DNA, 14, 8. https://doi.org/10.1186/s13100-023-00296-4
Nelson MG, Linheiro RS, Bergman CM (2017) McClintock: An integrated pipeline for detecting transposable element insertions in whole-genome shotgun sequencing data. G3: Genes, Genomes, Genetics, 7, 2763–2778. https://doi.org/10.1534/g3.117.043893
Rishishwar L, Mariño-Ramírez L, Jordan IK (2017) Benchmarking computational tools for polymorphic transposable element detection. Briefings in Bioinformatics, 18, 908–918. https://doi.org/10.1093/bib/bbw072
Vendrell-Mir P, Barteri F, Merenciano M, González J, Casacuberta JM, Castanera R (2019) A benchmark of transposon insertion detection tools using real data. Mobile DNA, 10, 53. https://doi.org/10.1186/s13100-019-0197-9
Verneret M, Le VA, Faraut T, Turpin J, Lerat E (2025) Particular sequence characteristics induce bias in the detection of polymorphic transposable element insertions. bioRxiv, ver. 4 peer-reviewed and recommended by PCI Genomics https://doi.org/10.1101/2024.09.25.614865