
OLIVA Adrien
- Unset, Unset, Unset, Australia
- Bioinformatics
Recommendations: 0
Review: 1
Review: 1
Estimating allele frequencies, ancestry proportions and genotype likelihoods in the presence of mapping bias
A novel genotype likelihood-based method to reduce mapping bias in low-coverage and ancient DNA studies
Recommended by Sebastian Ernesto Ramos-Onsins based on reviews by Maxime Lefebvre, Michael Westbury and Adrien OlivaThe study of genomic variability within and between populations, as well as among species, relies on comparative analyses of homologous positions—sites that share a common evolutionary origin. Homology is inferred through sequence similarity (Reeck et al. 1987). However, the ability to detect homologous regions can be compromised when sequence mismatches accumulate due to mutations, especially when analyzing short DNA fragments, as in short-read sequencing (Li et al. 2008). In the genomic era, accurately mapping homologous DNA fragments to a reference genome is essential for obtaining precise estimates of genetic variability and evolutionary inferences (e.g., Li et al. 2008; Ellegren 2014). However, short-read, high-throughput sequencing often introduces mapping bias, disproportionately favoring the reference allele. This bias distorts allele frequency estimates, ancestry proportions, and genotype likelihoods, impacting downstream analyses (e.g., Günther & Nettelblad 2019; Martiniano et al. 2020).
Mapping bias is particularly problematic in ancient DNA studies, where post-mortem damage exacerbates sequencing errors. DNA fragmentation limits read length, while deamination, causing G to A and C to U transitions, increases mismatches and further complicates homology identification (Dabney & Pääbo 2013). These degradation processes contribute to the misidentification of true variants, confounding evolutionary inferences. Various strategies have been developed to mitigate mapping bias, including the commonly used approach, called pseudo-haploid data, that randomly picks a single read at each analyzed position for each individual, thereby retaining a single allele at each polymorphic site (Günther & Nettelblad 2019; Barlow et al. 2020).
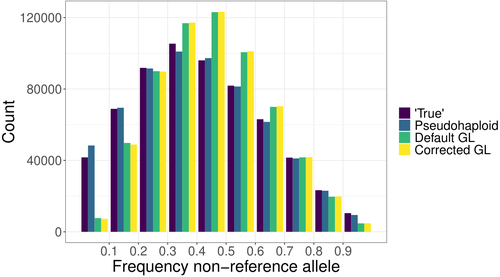
Günther et al. (2025) introduce a novel method to correct mapping bias using a genotype likelihood-based approach, incorporating a mapping bias ratio to adjust for reference allele overrepresentation. The method specifically targets known single nucleotide polymorphisms (SNPs) because in population genomic analysis of ancient DNA data, low coverage and post-mortem damage often hinder the ability to identify novel SNPs in most individuals. The analysis focuses on DNA fragmentation, assuming that deamination effects are minimal when considering ascertained SNPs. The proposed method was compared against existing approaches, including pseudo-haploid data and standard genotype likelihood-based probabilistic methods. The evaluation was performed using both empirical and simulated data. For empirical data, low-coverage sequencing data from the 1000 Genomes Project (Finnish in Finland, Japanese in Tokyo, Yoruba in Ibadan, Nigeria populations) was analyzed, while for simulated data, ancient DNA-like datasets were generated using ms-prime (Kelleher et al. 2016), modeling different sequencing depths, divergence times, and reference genome choices. The study assesses the impact of mapping bias on the ratio of reference versus non-reference allele mapping, the accuracy of SNP allele frequency estimates relative to true frequencies, the deviation and variance between estimated and true allele frequencies, population differentiation and the estimation of admixture proportions using supervised and unsupervised methods, considering both genotype likelihoods and genotype calls.
Günther et al. (2025) bring to light that all methods analyzed exhibit minor but systematic reference allele bias. The new corrected genotype likelihood method outperforms the standard genotype likelihood approach in correlating with true allele frequencies, although the pseudo-haploid method still provides the most accurate estimates. Mapping bias also affects ancestry estimation, leading to admixture proportion errors of up to 4%, though this effect is smaller than the 10% discrepancy observed across different inference methods.
The work performed by Günther et al. (2025) provides a rigorous and innovative evaluation of mapping bias in the context of ascertained SNPs, introducing a probabilistic approach that improves bias correction. Unlike non-probabilistic methods such as pseudo-haploid data, the genotype likelihood framework leverages all sequencing reads for each analyzed SNP, and can incorporate additional bias corrections, enhancing its applicability across different sequencing conditions. While probabilistic approaches offer clear advantages in bias correction, they can be less intuitive to interpret compared to traditional genotype calling methods. This study highlights that mapping bias is pervasive across all methods, influencing evolutionary inferences such as selection signals and population differentiation. Although the improvements in allele frequency recovery may seem modest, the genome-wide impact of mapping bias is significant, especially in ancient DNA studies, making bias correction essential for robust evolutionary analyses.
References
Barlow A, Hartmann S, Gonzalez J, Hofreiter M, Paijmans JLA. (2020) Consensify: A method for generating pseudohaploid genome sequences from palaeogenomic datasets with reduced error rates. Genes;11(1):50. https://doi.org/10.3390/genes11010050
Dabney J, Meyer M, Pääbo S. (2013) Ancient DNA damage. Cold Spring Harb Perspect Biol. 5(7):a012567. https://doi.org/10.1101/cshperspect.a012567
Ellegren H. (2014) Genome sequencing and population genomics in non-model organisms. Trends Ecol Evol. 29(1):51-63. https://doi.org/10.1016/j.tree.2013.09.008
Günther T, Nettelblad C. (2019) The presence and impact of reference bias on population genomic studies of prehistoric human populations. PLoS Genet.15(7):e1008302. https://doi.org/10.1371/journal.pgen.1008302
Günther T., Goldberg A., Schraiber J. G. (2025) Estimating allele frequencies, ancestry proportions and genotype likelihoods in the presence of mapping bias. bioRxiv, ver. 5 peer-reviewed and recommended by PCI Genomics https://doi.org/10.1101/2024.07.01.601500
Kelleher J., Etheridge A. M., McVean G. (2016) Efficient coalescent simulation and genealogical analysis for large sample sizes. PLoS computational biology, 12(5):e1004842. https://doi.org/10.1371/journal.pcbi.1004842
Li H, Ruan J, Durbin R. (2008) Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 18(11):1851-8. https://doi.org/10.1101/gr.078212.108
Reeck GR, de Haën C, Teller DC, Doolittle RF, Fitch WM, Dickerson RE, et al. (1987) "Homology" in proteins and nucleic acids: a terminology muddle and a way out of it. Cell. 50 (5): 667. https://doi.org/10.1016/0092-8674(87)90322-9