
Unveiling transposon dynamics: Advancing TE expression analysis in Drosophila with long-read sequencing
Identification and quantification of transposable element transcripts using Long-Read RNA-seq in Drosophila germline tissues
Abstract
Recommendation: posted 02 August 2024, validated 06 August 2024
Pollet, N. (2024) Unveiling transposon dynamics: Advancing TE expression analysis in Drosophila with long-read sequencing. Peer Community in Genomics, 100250. 10.24072/pci.genomics.100250
Recommendation
Transposable elements (TEs) are mobile genetic elements with an intrinsic mutagenic potential that influences the physiology of any cell type, whether somatic or germinal. Measuring TE expression is a fundamental prerequisite for analysing the processes leading to the activity of TE-derived sequences. This applies to both old and recent TEs, as even if they are deficient in mobilisation, transcription of TE sequences alone can impact neighbouring gene expression and other cellular activities.
In terms of TE physiology, transcription is crucial for mobilisation activity. The transcription of some TEs can be tissue-specific and associated with splicing events, as exemplified by the P-element isoforms in the fruit fly (Laski et al. 1986). Regarding host cell physiology, TE transcripts can include nearby exons, with or without splicing, and such chimeric transcripts can significantly alter gene activity. Thus, quantitative and qualitative analyses must be conducted to assess TE function and how they can modify genomic activities. Yet, due to the polymorphic, interspersed, and repetitive nature of TE sequences, the quantitative and qualitative analysis of TE transcript levels using short-read sequencing remains challenging (Lanciano and Cristofari 2020).
In this context, Rebollo et al. (2024) employed nanopore long-read sequencing to analyse cDNAs derived from Drosophila melanogaster germline RNAs. The authors constructed two long-read cDNA libraries from pooled ovaries and testes using a protocol to obtain full-length cDNAs and sequenced them separately. They carefully compared their results with their short-read datasets. Overall, their observations corroborate known patterns of germline-specific expression of certain TEs and provide initial evidence of novel spliced TE transcript isoforms in Drosophila.
Rebollo and colleagues have provided a well-documented and detailed analysis of their results, which will undoubtedly benefit the scientific community. They presented the challenges and limitations of their approach, such as the length of the transcripts, and provided a reproducible analysis workflow that will enable better characterisation of TE expression using long-read technology.
Despite the small number of samples and limited sequencing depth, this pioneering study strikingly demonstrates the potential of long-read sequencing for the quantitative and qualitative analysis of TE transcription, a technology that will facilitate a better understanding of the transposon landscape.
References
Lanciano S, Cristofari G (2020) Measuring and interpreting transposable element expression. Nature Reviews Genetics, 21, 721–736. https://doi.org/10.1038/s41576-020-0251-y
Laski FA, Rio DC, Rubin GM (1986) Tissue specificity of Drosophila P element transposition is regulated at the level of mRNA splicing. Cell, 44, 7–19. https://doi.org/10.1016/0092-8674(86)90480-0
Rebollo R, Gerenton P, Cumunel E, Mary A, Sabot F, Burlet N, Gillet B, Hughes S, Oliveira DS, Goubert C, Fablet M, Vieira C, Lacroix V (2024) Identification and quantification of transposable element transcripts using Long-Read RNA-seq in Drosophila germline tissues. bioRxiv, ver.4 peer-reviewed and recommended by PCI Genomics. https://doi.org/10.1101/2023.05.27.542554

The recommender in charge of the evaluation of the article and the reviewers declared that they have no conflict of interest (as defined in the code of conduct of PCI) with the authors or with the content of the article. The authors declared that they comply with the PCI rule of having no financial conflicts of interest in relation to the content of the article.
This work was supported by French National research agency (ANR project ANR-16-CE23-0001 502 ‘ASTER and ANR-20-CE02-0015 LongevitY), along with UDL-FAPESP2020.
Evaluation round #2
DOI or URL of the preprint: https://doi.org/10.1101/2023.05.27.542554
Version of the preprint: 3
Author's Reply, 12 Jul 2024
Decision by Nicolas Pollet, posted 20 Jan 2024, validated 23 Jan 2024
Dear Rita Rebollo,
The three reviewers have made available their comments on your revised manuscript. The referees' comments indicate that they are very satisfied with the modifications made. Yet, one reviewer has critical and well-argued comments that need to be answered in detail because they can potentially prevent recommendation.
I would especially invite you to answer the points raised on novel spliced TE isoforms :
- TE insertions refer to recent insertions mediated by a functional TE
- TE annotation impact on your analysis
- discuss the impact of chimeric cDNA (most likely derived from coligation, something that is well known but insufficiently quantified) on your results. Possibly trying to figure out a quantification of chimeric cDNAs based on easily recognizable events from long reads derived from abundant transcripts.
I will send you the two tables mentioned by the reviewer in a separate e-mail.
With best wishes,
Nicolas Pollet
Reviewed by anonymous reviewer 1, 11 Jan 2024
Rebollo et al., Identification and quantification of transposable element transcripts using Long-Read RNA-seq in Drosophila germline tissues.
– revised manuscript
The authors have put substantial effort in revising the manuscript, which, in my opinion, is now significantly improved. They have addressed sufficiently all my comments, especially by clarifying the issue of single- vs multi-mapping TE reads and by softening their conclusions on the biological significance of the non-replicated data. Thus, I maintain my feeling, that the strength of the manuscript lays in the technology used and the developed analysis tools. These, largely thanks to the comments of the other two reviewers, should now be much more reproducible and merit their sharing with the community.
Below, I have suggested a few last modifications:
2, 252, 266, 303 and throughout: For clarity, in a final version of the manuscript, the authors should better avoid using “long-read RNA-seq” and replace with “long-read cDNA-seq”, in order to avoid any confusion with ONT direct RNA sequencing approaches.
258: The statement “between ~1 to ~3 million reads per tissue” when there are only two samples in total (one per tissue) is misleading. Please rewrite.
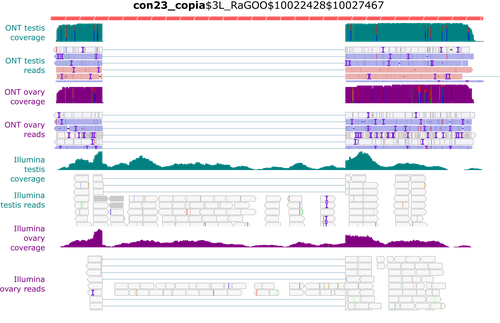
Fig 1A and S10-11: I strongly feel that the transcript length bias, which is well explained in the text, is very important for this study and for others that might want to perform similar type of analysis. Thus, graphs from figures S10 and S11 should be moved to the main fig 1 (either in addition to or replacing the panel 1A).
288: Replace “to ensure” with “to check if”.
294: Move “(as suggested by the cDNA profile, Figure S1)” at the end of the sentence.
349: Please add a caution note reminding the reader that definite conclusions on the difference between sexes would require replicating the results.
371: Suggestion: replace “single-copy” with “copy-specific”.
393: Should be: “unambiguously mapping”
Fig 3B: The y-axis range for Pogo element graphs is too high. Add: “Each dot represents a unique genomic copy”.
487: Should be: “ONT long-read sequencing detects”
488: Please add that, knowing the poor recovery of long transcripts, expression of longer TEs copies might be underestimated. This statement, present in lines 510-512, can be moved up.
501: Please change to “may unvail” (in regard to lack of replicates, frequent low read support and more extensive splicing analysis).
544: Should be: “only one or two”
602: Should be: “to specific copies of transposable elements”.
616: Should be: “retrotransposed”
610: Should be: “TE transcripts are spliced”
614: Please add: “While our results suggest that TE splicing could be prevalent, additional studies with biological replicates, high sequencing coverage and mechanistic insights into the splicing machinery will be needed to confirm our observations.” Or a similar statement.
Download the review
Reviewed by Christophe Antoniewski, 13 Jan 2024
First of all, my apologies to the authors for taking a long time to re-evaluate the manuscript.
I have read the revised version in detail and find that the authors have done an excellent job and addressed the vast majority of my comments satisfactorily. I particularly appreciate the effort on rewriting the methods and depositing them in a GitLab repository.
I think that the article is now reproducible and above all that it can be useful to a large community of biologists, well beyond Drosophila researchers, including researchers working in human genomics.
It was worth it, wasn't it?
Reviewed by Silke Jensen , 10 Jan 2024
, 10 Jan 2024
The supplementary excel files that are cited in the review document, destined to the authors, will be sent by e-mail.
Download the reviewEvaluation round #1
DOI or URL of the preprint: https://doi.org/10.1101/2023.05.27.542554
Version of the preprint: 2
Author's Reply, 07 Dec 2023
Decision by Nicolas Pollet, posted 14 Aug 2023, validated 14 Aug 2023
Dear Rita Rebollo and colleagues,
Your manuscript entitled "Identification and quantification of transposable element transcripts using Long-Read RNA-seq in Drosophila germline tissues" has been reviewed by three colleagues. Globally the reviews are of high quality and positive, and find that your study has many merits, but there are substantial and major criticisms that you need to adress before I can finally decide on wether this preprint can be recommended or not.
Especially, I would like to underscore the availability of the data used in your analysis and the availability of re-usable bioinformatics methods.
With my best wishes,
Nicolas Pollet
Reviewed by anonymous reviewer 1, 18 Jul 2023
In the proposed manuscript, Rebollo et al explore the use of the long-read Oxford Nanopore (ONT) sequencing technology to describe qualitatively and quantitatively the transcriptional landscape of transposable elements in the Drosophila gonads. The authors generate, sequence and analyze long-read cDNA libraries (one replicate from each tissue type), as well as compare the obtained results to previously published short-read datasets.
The manuscript is timely, as we can observe a growing interest in the use of ONT technology for transcriptome analysis, especially in the field of DNA repeats. As such, this work will certainly be useful for many researchers, even more so, that it includes clear explanations of all wet-lab and data analysis approaches. The manuscript exposes clearly the strengths, as well the limitations of the techniques used. The manuscript is also quite unique in its detailed comparison between short- and long-read transcriptomic datasets, which shows how these two technologies can complement each other on different levels.
The manuscript could benefit from the following improvements or clarifications:
Major comments:
Regarding “unique” and “unique best” mapping reads vs multi-mappers:
1- According to the text and the Table S1, the “unique best” mapping reads represent 91% and 99% of the sequenced libraries. This however, relates to total reads, only small percentage of which maps to TEs. Thus, effectively, this percentage is true for genes. What are the fractions of “unique” and “unique best” reads for TE-mapping reads only? Supposedly, these could be different than for single copy genes. How does this compare with short-read libraries? To which extend long-reads reduce or overcome the multimapping issue? This would be interesting to show clearly and it is also important for the downstream analysis.
If there is a significant fraction of multi-mapping reads among all TE-mapping reads, are these reads taken under account in transcript abundance quantifications? If present, these multi-mappers should contribute to the family-level transcript estimates (Fig 2), and should be taken under account when quantifying copy-specific expression (Fig 3 and 4). If, for example, for a given TE family, there are significantly more multi-mapping reads than unique best aligners, any conclusions on how all the copies contribute the total transcript levels would be impossible to make. The analysis of expressed vs non-expressed TE copies would still hold true, but only for genomic loci that present enough sequence variation to produce transcripts with unique best alignments.
The authors should clarify this by providing the statistics of multi-mapped reads for TEs, performing any additional analysis if necessary and adjusting their conclusions if required.
2 -The above point brings up the question of sequence variation between genomic copies of different expressed TE families, which, in the current version of the manuscript, is not much discussed. Expression of evolutionary younger TEs, with lower sequence divergence, would obviously be more difficult to quantify. This would be particularly relevant for the search of full-length TE transcripts (Fig 4), which would carry less informative sequence variation. The authors could include sequence variation of genomic copies (as they do for sequence variants of transcripts in Fig 5) in their analysis or, minimally, they should comment on the limitations that could be related to the potential lack of such variation. Again, this issue will be less relevant if no (or very few) TE-specific multi-mappers are in fact found in the libraries.
Regarding mapping reads to features:
3– A substantial number of TEs is located in intronic sequences. Taking under account how the authors assign reads to features, would intronic TEs in expressed genes be taken under account or omitted? This is not clear. Theoretically, in such cases, both features (the gene and the intronic TE) could be fully covered. In other words, are TEs belonging to the “intron” category if Fig 1E found only (or primarily) in non-expressed genes?
Table S1 and line 215:
Please add read length statistics (median, N50) separately for both samples. Read length will influence some of the downstream analysis, thus it would be important to indicate it.
Additional minor comments:
Methods:
Please specify how much total RNA was used as input for the TeloPrime cDNA amplification.
Lines 238-240:
The use of percent ranges (e.g. 37-48%) is misleading, when in fact only two samples are analyzed. Replacing with the two obtained values only, would be more accurate.
Fig 1B and D:
Transcript coverage in Fig 1B should be plotted as a function of transcript length, similarly as done for the figure panel 1D.
Related to the above point (lines 229-230 and Fig 1D), the text of the results sections should not omit the fact that good correlation is achieved only for short transcripts. Although this detailed explanation is coming later in the text, it would be easier for the reader, if the point of underrepresentation of long transcripts was clarified up front.
Fig 1C:
Please correct, testis > testes
Line 246-247 and Fig 1F:
The authors to some extend contrast TE transcripts with gene transcripts by stating: “on the other hand, gene transcripts may reach 5kb”. Although this is true based on the data, it should be taken under account that overall genes are much more highly expressed than TEs, increasing the chance of detection of underrepresented long transcripts. If the authors wish to make such comparison, they should include transcript abundance as a contributing factor.
Overall, due to lack of replicates and important difference on coverage, any conclusions regarding comparison between samples should be made very carefully. The authors do acknowledge this and mostly remain careful in their conclusions (e.x. line 293-294). However, throughout the paragraph (lines 279-294) the authors should avoid direct comparisons of read numbers between female and male libraries. Without any kind of normalization statements such as “but both families with higher transcripts in males” are not very meaningful. Also, regarding the observation that the global proportion of reads mapping to TEs is significantly higher in testes than in ovaries, it was not clear from the text, if this was also true for the previously published data (Larat et al 2017). If so, this would strengthen the obtained result, as it does in Fig 2C for the expression at the family level.
Finally, are TEs with male-specific transcripts (HETA, TAHRE) enriched on the Y chromosome?
Fig 3A:
Please unify: “copy number” = “# of genomic copies”
Line 360:
Please remove the abbreviation “v.r.t.”
Line 356 and the results section below:
In light of the demonstration that the technical approach taken is strongly underestimating very long transcripts (Fig 1), the section title should be toned down to “are rarely detected” rather than “rarely transcribed”. Also, the authors should remind the reader of this technical limitation here and tone down their conclusion as to whether the detected transcripts are fully reflecting the transcripts presents in the tissues investigated.
Fig 5:
Please enlarge fonts for TE family names
Download the review



