
Latest recommendations

| Id | Title | Authors | Abstract | Picture | Thematic fields | Recommender | Reviewers | Submission date▼ | |
|---|---|---|---|---|---|---|---|---|---|
09 Oct 2020
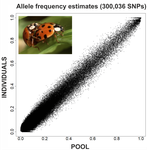
An evaluation of pool-sequencing transcriptome-based exon capture for population genomics in non-model speciesAssessing a novel sequencing-based approach for population genomics in non-model speciesRecommended by Thomas Derrien and Sebastian E. Ramos-Onsins based on reviews by Valentin Wucher and 1 anonymous reviewerDeveloping new sequencing and bioinformatic strategies for non-model species is of great interest in many applications, such as phylogenetic studies of diverse related species, but also for studies in population genomics, where a relatively large number of individuals is necessary. Different approaches have been developed and used in these last two decades, such as RAD-Seq (e.g., Miller et al. 2007), exome sequencing (e.g., Teer and Mullikin 2010) and other genome reduced representation methods that avoid the use of a good reference and well annotated genome (reviewed at Davey et al. 2011). However, population genomics studies require the analysis of numerous individuals, which makes the studies still expensive. Pooling samples was thought as an inexpensive strategy to obtain estimates of variability and other related to the frequency spectrum, thus allowing the study of variability at population level (e.g., Van Tassell et al. 2008), although the major drawback was the loss of information related to the linkage of the variants. In addition, population analysis using all these sequencing strategies require statistical and empirical validations that are not always fully performed. A number of studies aiming to obtain unbiased estimates of variability using reduced representation libraries and/or with pooled data have been performed (e.g., Futschik and Schlötterer 2010, Gautier et al. 2013, Ferretti et al. 2013, Lynch et al. 2014), as well as validation of new sequencing methods for population genetic analyses (e.g., Gautier et al. 2013, Nevado et al. 2014). Nevertheless, empirical validation using both pooled and individual experimental approaches combined with different bioinformatic methods has not been always performed. References [1] Choquet et al. (2019). Towards population genomics in non-model species with large genomes: a case study of the marine zooplankton Calanus finmarchicus. Royal Society open science, 6(2), 180608. doi: https://doi.org/10.1098/rsos.180608 | An evaluation of pool-sequencing transcriptome-based exon capture for population genomics in non-model species | Emeline Deleury, Thomas Guillemaud, Aurélie Blin & Eric Lombaert | <p>Exon capture coupled to high-throughput sequencing constitutes a cost-effective technical solution for addressing specific questions in evolutionary biology by focusing on expressed regions of the genome preferentially targeted by selection. Tr... | | Bioinformatics, Population genomics | Thomas Derrien | 2020-02-26 09:21:11 | ||
06 Apr 2021
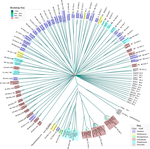
Evidence for shared ancestry between Actinobacteria and Firmicutes bacteriophagesViruses of bacteria: phages evolution across phylum boundariesRecommended by Denis Tagu based on reviews by 3 anonymous reviewersBacteria and phages have coexisted and coevolved for a long time. Phages are bacteria-infecting viruses, with a symbiotic status sensu lato, meaning they can be pathogenic, commensal or mutualistic. Thus, the association between bacteria phages has probably played a key role in the high adaptability of bacteria to most - if not all – of Earth’s ecosystems, including other living organisms (such as eukaryotes), and also regulate bacterial community size (for instance during bacterial blooms). As genetic entities, phages are submitted to mutations and natural selection, which changes their DNA sequence. Therefore, comparative genomic analyses of contemporary phages can be useful to understand their evolutionary dynamics. International initiatives such as SEA-PHAGES have started to tackle the issue of history of phage-bacteria interactions and to describe the dynamics of the co-evolution between bacterial hosts and their associated viruses. Indeed, the understanding of this cross-talk has many potential implications in terms of health and agriculture, among others. The work of Koert et al. (2021) deals with one of the largest groups of bacteria (Actinobacteria), which are Gram-positive bacteria mainly found in soil and water. Some soil-born Actinobacteria develop filamentous structures reminiscent of the mycelium of eukaryotic fungi. In this study, the authors focused on the Streptomyces clade, a large genus of Actinobacteria colonized by phages known for their high level of genetic diversity. The authors tested the hypothesis that large exchanges of genetic material occurred between Streptomyces and diverse phages associated with bacterial hosts. Using public datasets, their comparative phylogenomic analyses identified a new cluster among Actinobacteria–infecting phages closely related to phages of Firmicutes. Moreover, the GC content and codon-usage biases of this group of phages of Actinobacteria are similar to those of Firmicutes. This work demonstrates for the first time the transfer of a bacteriophage lineage from one bacterial phylum to another one. The results presented here suggest that the age of the described transfer is probably recent since several genomic characteristics of the phage are not fully adapted to their new hosts. However, the frequency of such transfer events remains an open question. If frequent, such exchanges would mean that pools of bacteriophages are regularly fueled by genetic material coming from external sources, which would have important implications for the co-evolutionary dynamics of phages and bacteria. References Koert, M., López-Pérez, J., Courtney Mattson, C., Caruso, S. and Erill, I. (2021) Evidence for shared ancestry between Actinobacteria and Firmicutes bacteriophages. bioRxiv, 842583, version 5 peer-reviewed and recommended by Peer community in Genomics. doi: https://doi.org/10.1101/842583 | Evidence for shared ancestry between Actinobacteria and Firmicutes bacteriophages | Matthew Koert, Júlia López-Pérez, Courtney Mattson, Steven M. Caruso, Ivan Erill | <p>Bacteriophages typically infect a small set of related bacterial strains. The transfer of bacteriophages between more distant clades of bacteria has often been postulated, but remains mostly unaddressed. In this work we leverage the sequencing ... | | Evolutionary genomics | Denis Tagu | 2019-12-10 15:26:31 | ||
24 Sep 2020

A rapid and simple method for assessing and representing genome sequence relatednessA quick alternative method for resolving bacterial taxonomy using short identical DNA sequences in genomes or metagenomesRecommended by B. Jesse Shapiro based on reviews by Gavin Douglas and 1 anonymous reviewerThe bacterial species problem can be summarized as follows: bacteria recombine too little, and yet too much (Shapiro 2019). References Arevalo P, VanInsberghe D, Elsherbini J, Gore J, Polz MF (2019) A Reverse Ecology Approach Based on a Biological Definition of Microbial Populations. Cell, 178, 820-834.e14. https://doi.org/10.1016/j.cell.2019.06.033 | A rapid and simple method for assessing and representing genome sequence relatedness | M Briand, M Bouzid, G Hunault, M Legeay, M Fischer-Le Saux, M Barret | <p>Coherent genomic groups are frequently used as a proxy for bacterial species delineation through computation of overall genome relatedness indices (OGRI). Average nucleotide identity (ANI) is a widely employed method for estimating relatedness ... | | Bioinformatics, Metagenomics | B. Jesse Shapiro | Gavin Douglas | 2019-11-07 16:37:56 |




