
TIRERA Sourakhata
Recommendations: 0
Review: 1
Review: 1
mbctools: A User-Friendly Metabarcoding and Cross-Platform Pipeline for Analyzing Multiple Amplicon Sequencing Data across a Large Diversity of Organisms
One tool to metabarcode them all
Recommended by Nicolas Pollet based on reviews by Ali Hakimzadeh and Sourakhata TireraOne way to identify all organisms at their various life stages is by their genetic signature. DNA-based taxonomy, gene tagging and barcoding are different shortcuts used to name such strategies (Lamb et al. 2019; Tautz et al. 2003). Reading and analyzing nucleic acid sequences to perform genetic inventories is now faster than ever, and the latest nucleic acid sequencing technologies reveal an impressive taxonomic, genetic, and functional diversity hidden in all ecosystems (Lamb et al. 2019; Sunagawa et al. 2015). This knowledge should enable us to evaluate biodiversity across its scales, from genetic to species to ecosystem and is sometimes referred to with the neologism of ecogenomics (Dicke et al. 2004).
The metabarcoding approach is a key workhorse of ecogenomics. At the core of metabarcoding strategies lies the sequencing of amplicons obtained from so-called multi-template PCR, a formidable and potent experiment with the potential to unravel hidden biosphere components from different samples obtained from organisms or the environment (Kalle et al. 2014; Rodríguez-Ezpeleta et al. 2021). Next to this core approach, and equally important, lies the bioinformatic analysis to convert the raw sequencing data into amplicon sequence variants or operational taxonomic units and interpretable abundance tables.
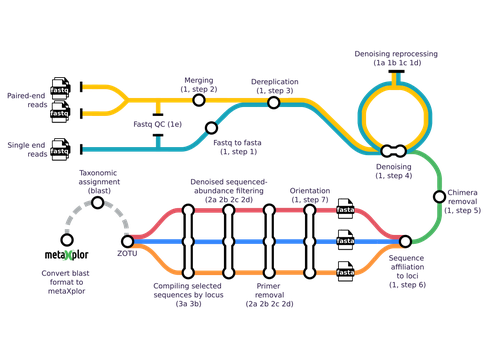
Methodologically, the analysis of sequences obtained from metabarcoding projects is replete with devilish details. This is why different pipelines and tools have been developed, starting with mothur (Schloss et al. 2009) and QIIME 2 (Bolyen et al. 2019), but including more user friendly tools such as FROGS (Escudié et al. 2018). Yet, across all available tools, scientists must choose the optimal algorithms and parameter values to filter raw reads, trim primers, identify chimeras and cluster reads into operational taxonomic units. In addition, the number of genetic markers used to characterize a sample using metabarcoding has increased as sequencing methods are now less costly and more efficient. In such cases, results and interpretations may become limited or confounded. This is where the novel tools proposed by Barnabé and colleagues (2024), mbctools, will benefit researchers in this field.
The authors provide a detailed description with a walk-through of the mbctools pipeline to analyse raw reads obtained in a metabarcoding project. The mbctools pipeline can be installed under different computing environments, requires only VSEARCH and a few Python dependencies, and is easy to use with a menu-driven interface. Users need to prepare their data following simple rules, providing single or paired-end reads, primer and target database sequences. An interesting feature of mbctools output is the possibility of integration with the metaXplor visualization tool developed by the authors (Sempéré et al. 2021). As it stands, mbctools should be used for short-read sequences. The taxonomy assignment module has the advantage to enable parameters exploration in an easy way, but it may be oversimplistic for specific taxa.
The lightweight aspect of mbctools and its overall simplicity are appealing. These features will make it a useful pipeline for training workshops and to help disseminate the use of metabarcoding. It also holds the potential for further improvement, by the developers or by others. In the end, mbctools will support study reproducibility by enabling a streamlined analysis of raw reads, and like many useful tools, only time will tell whether it is widely adopted.
References
Barnabé C, Sempéré G, Manzanilla V, Millan JM, Amblard-Rambert A, Waleckx E (2024) mbctools: A user-friendly metabarcoding and cross-platform pipeline for analyzing multiple amplicon sequencing data across a large diversity of organisms. bioRxiv, ver. 2 peer-reviewed and recommended by PCI Genomics https://doi.org/10.1101/2024.02.08.579441
Bolyen E, Rideout JR, Dillon MR, Bokulich NA, et al. (2019) Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nature Biotechnology, 37, 852–857. https://doi.org/10.1038/s41587-019-0209-9
Dicke M, van Loon JJA, de Jong PW (2004) Ecogenomics benefits community ecology. Science, 305, 618–619. https://doi.org/10.1126/science.1101788
Escudié F, Auer L, Bernard M, Mariadassou M, Cauquil L, Vidal K, Maman S, Hernandez-Raquet G, Combes S, Pascal G (2018) FROGS: Find, Rapidly, OTUs with Galaxy Solution. Bioinformatics, 34, 1287-1294. https://doi.org/10.1093/bioinformatics/btx791
Kalle E, Kubista M, Rensing C (2014) Multi-template polymerase chain reaction. Biomolecular Detection and Quantification, 2, 11–29. https://doi.org/10.1016/j.bdq.2014.11.002
Lamb CT, Ford AT, Proctor MF, Royle JA, Mowat G, Boutin S (2019) Genetic tagging in the Anthropocene: scaling ecology from alleles to ecosystems. Ecological Applications, 29, e01876. https://doi.org/10.1002/eap.1876
Rodríguez-Ezpeleta N, Zinger L, Kinziger A, Bik HM, Bonin A, Coissac E, Emerson BC, Lopes CM, Pelletier TA, Taberlet P, Narum S (2021) Biodiversity monitoring using environmental DNA. Molecular Ecology Resources, 21, 1405–1409. https://doi.org/10.1111/1755-0998.13399
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Applied and Environmental Microbiology 75, 7537-41. https://doi.org/10.1128/AEM.01541-09
Sempéré G, Pétel A, Abbé M, Lefeuvre P, Roumagnac P, Mahé F, Baurens G, Filloux D 2021 metaXplor: an interactive viral and microbial metagenomic data manager. Gigascience, 10, https://doi.org/10.1093/gigascience/giab001
Sunagawa S, Coelho LP, Chaffron S, Kultima JR, Labadie K, Salazar G, Djahanschiri B, Zeller G, Mende DR, Alberti A, Cornejo-Castillo FM, Costea PI, Cruaud C, d’Ovidio F, Engelen S, Ferrera I, Gasol JM, Guidi L, Hildebrand F, Kokoszka F, Lepoivre C, Lima-Mendez G, Poulain J, Poulos BT, Royo-Llonch M, Sarmento H, Vieira-Silva S, Dimier C, Picheral M, Searson S, Kandels-Lewis S, Tara Oceans coordinators, Bowler C, de Vargas C, Gorsky G, Grimsley N, Hingamp P, Iudicone D, Jaillon O, Not F, Ogata H, Pesant S, Speich S, Stemmann L, Sullivan MB, Weissenbach J, Wincker P, Karsenti E, Raes J, Acinas SG, Bork P (2015) Structure and function of the global ocean microbiome. Science, 348, 1261359. https://doi.org/10.1126/science.1261359
Tautz D, Arctander P, Minelli A, Thomas RH, Vogler AP (2003) A plea for DNA taxonomy. Trends in Ecology & Evolution, 18, 70–74. https://doi.org/10.1016/S0169-5347(02)00041-1