
Leveraging HHpred with rigorous validation for improved detection of host-virus homologies
 based on reviews by 2 anonymous reviewers
based on reviews by 2 anonymous reviewers
Re-annotation of SARS-CoV-2 proteins using an HHpred-based approach opens new opportunities for a better understanding of this virus
Abstract
Recommendation: posted 09 November 2024, validated 13 November 2024
Narayan, J. (2024) Leveraging HHpred with rigorous validation for improved detection of host-virus homologies. Peer Community in Genomics, 100247. 10.24072/pci.genomics.100247
Recommendation
The assessment by Brézellec (2024) of the quality of HHpred-based SARS-CoV-2 protein annotations against the traditional Pfam annotations is highly justified and valuable. HHpred’s ability to detect remote homologies offers an expanded view of viral protein similarities, potentially uncovering subtle functional mimicries that Pfam may miss due to its sensitivity limitations when dealing with divergent sequences. However, the accuracy and specificity of HHpred results can be compromised by false positives, especially when dealing with complex viral proteins that feature transmembrane or low-complexity regions prone to spurious matches.
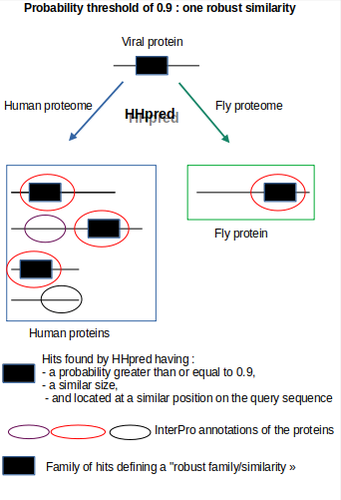
To address this, the author made a thoughtful decision to implement a multi-step validation protocol. This approach included establishing progressively lower probability thresholds to capture weaker but biologically plausible hits, and organizing hits into “families” of similarly located alignments to validate the robustness of matches. They also cross-verified results by running SARS-CoV-2 protein queries against non-human proteomes (plants, fruit flies, bacteria, and archaea), allowing them to discern between biologically meaningful matches and potentially random alignments. By adding manual verification with InterPro domain annotations, the authors took additional steps to ensure that identified similarities were not only statistically significant but also biologically relevant.
This rigorous validation strategy adds a layer of reliability to HHpred results, demonstrating an effective maximization of sensitivity while maintaining specificity. This approach yielded biologically intriguing and previously undocumented similarities, such as between the Spike-prominin and ORF3a-GPCR, underscoring the quality and depth of the annotation process. These findings highlight a pathway for further experimental validation and illustrate the potential of HHpred to contribute high-quality insights when applied with careful quality control measures.
In summary, the decision to adopt HHpred (Gabler et al. 2020) and enhance its outputs with a robust quality validation process not only improved the depth of SARS-CoV-2 protein annotations but also established a high standard for future viral annotation projects, striking an effective balance between discovery potential and annotation quality. The authors have conducted a study that is methodologically rigorous, well-detailed, and highly pertinent to the field. This work stands as a significant contribution to the scientific community, providing resources and insights that are likely to guide future research in this area.
References
Brézellec, P (2024) Re-annotation of SARS-CoV-2 proteins using an HHpred-based approach opens new opportunities for a better understanding of this virus. bioRxiv, ver. 3 peer-reviewed and recommended by PCI Genomics. https://doi.org/10.1101/2023.06.06.543855
Gabler F, Nam S-Z, Till S, Mirdita M, Steinegger M, Söding J, Lupas AN, Alva V (2020) Protein Sequence Analysis Using the MPI Bioinformatics Toolkit. Current Protocols in Bioinformatics, 72, e108. https://doi.org/10.1002/cpbi.108

The recommender in charge of the evaluation of the article and the reviewers declared that they have no conflict of interest (as defined in the code of conduct of PCI) with the authors or with the content of the article. The authors declared that they comply with the PCI rule of having no financial conflicts of interest in relation to the content of the article.
The author declares that he has received no specific funding for this study
Evaluation round #2
DOI or URL of the preprint: https://www.biorxiv.org/content/10.1101/2023.06.06.543855v2
Version of the preprint: 1
Author's Reply, 19 Sep 2024
Dear Editor,
Thank you very much for your comments! They really made me think, and I believe I've found a way to strengthen my arguments even more.
Before addressing the different points you raised (section 2) and presenting other compelling arguments supporting my results (section 3), I will clarify some points (section 1). Finally (section 4), I will conclude that thanks to the suggestions and comments provided by you and the referees, I am confident that my paper now presents reliable and robust scientific findings.
--> 1./ To provide a clearer understanding of my work, I will highlight the following two key points:
First, employing the methodology used in the paper allows us to recover previously known results about SARS-CoV-2 sequences, as evidenced in the section titled "A 'highly robust' or 'very robust' similarity, already documented in literature, was detected on the following 4 proteins." This lends credibility to our methodology.
Second, a part of this methodology is based on an idea described in (Gabler et al., 2020) which is not yet taken into account in HHpred (see HHPRED documentation section "Understanding Results" subsection "Check if you can reproduce the results with other parameters"):
To assess the robustness of my HHpred results, I conducted the query against additional proteomes, including Arabidopsis thaliana, Drosophila melanogaster, Escherichia coli, and Haloferax volcanii (detailed in the section "Procedure for assessing the robustness of HHpred results"). This cross-species analysis allowed me to identify and exclude homologies that might be specific to the human proteome. For instance, any matches with a probability greater than 0.95 that were not replicated in at least one of these other organisms were considered inconclusive and omitted from my analysis. By prioritizing data supported by evidence from multiple proteomes, I aimed to enhance the reliability of my findings.
--> 2. Answers to the questions you raised and suggestions you made:
2.1./ "One of their primary claims is that the SARS-CoV-2 Spike protein is similar to the prominin domain of a human protein. However, this conclusion is based solely on profile-based searches, which compare sequence profiles rather than employing more robust structural or functional analyses. This approach may be considered somewhat crude and may not provide reliable or accurate insights into protein function or interactions (1,2,3)."
As you noted, I primarily relied on HHPRED for my analysis. However, prior to using HHPRED, I attempted to reannotate SARS-CoV-2 proteins using Phyre2 (Kelley et al., 2015), but this approach did not yield any significant results.
To address a reviewer's suggestion, I further explored cross-validation by employing AlphaFold databases and FoldSeek (van Kempen et al., 2023). Despite these efforts, I was unable to uncover any additional insights.
Due to several factors, I've chosen not to include these unsuccessful attempts in my paper.
2.2./ "Additionally, the paper's second major conclusion involves the ORF3a protein of SARS-CoV-2. The authors themselves describe this finding as non-significant, raising further concerns about the robustness of their overall methodology and the validity of their conclusions"
The conclusions you're referring to are about ORF3a of SARS-CoV-1, not SARS-CoV-2 (keep in mind that this work is devoted to SARS-CoV-2). For the ORF3a protein expressed by SARS-CoV-1, I am indeed unable to draw any conclusions based on my methodology. However, the results concerning ORF3a of SARS-CoV-2 are reliable, according to the methodology I employed.
2.3./ "I recommend using multiple computational tools and databases for homology detection and structure prediction, such as PSI-BLAST and SWISS-MODEL, to cross-validate the findings from HHpred. "
Yes, you're right. However, as mentioned above, I tried other tools. In addition, it is now well-documented that profile HMMs outperform sequence profiles (e.g., PSI-BLAST) in the detection of remote homologs and in the quality of alignments (Söding, 2005). Regarding SWISS-MODEL, I feel (but I may be wrong) that it is, at best, somewhat redundant with AlphaFold results.
2.4./ "Additionally, conducting phylogenetic analysis to trace the evolutionary relationships between SARS-CoV-2 proteins and their human counterparts can provide further context on the functional and structural conservation of these proteins across different species. It is also advisable to use co-evolution analysis tools like GREMLIN or EVcouplings to identify co-evolving residues that might indicate functional interactions between SARS-CoV-2 proteins and human proteins."
You raised very interesting points that are unfortunately beyond my expertise. I am unable to explore these topics further. However, note that, to the best of my knowledge, I believe AlphaFold detects and takes into account the co-evolution of residues.
--> 3./ Convincing evidence supporting our findings (I included an extended version of what follows in the last version of the paper, see section Further investigation of already found similarities):
Recall that we demonstrated that: 1./ SARS-CoV-2 Spike S shares similarities with Human Prominin 1, and 2./ SARS-CoV-2 ORF3a shares similarities with several Human G-coupled proteins, with "Lutropin-choriogonadotropic hormone receptor" as the closest match.
To further investigate these similarities, we used HHPRED to compare Prominin 1 and Lutropin-choriogonadotropic hormone receptor to a set of viral proteins.
Interestingly, Prominin 1 showed similarities with 6 viral Spike glycoproteins, primarily from coronaviruses. Equally interestingly, Lutropin-choriogonadotropic hormone receptor showed similarities with 23 viral G-protein coupled receptors, particularly from Herpesvirales.
The overall process can be viewed as a reciprocal best hit (or bidirectional best hit): 1. a part of SARS-CoV-2 Spike S is similar to a part of human Prominin 1, which is itself similar to several viral Spike glycoproteins; 2. a part of SARS-CoV-2 ORF3a is similar to a part of a human G-coupled protein, which is also itself similar to several viral G-coupled proteins.
--> 4. Conclusion:
Thanks to your valuable feedback and the reviewers' comments, I am confident that this paper now meets rigorous scientific standards. I am convinced that our main finding — the similarity between the SARS-CoV-2 Spike protein and Human Prominin 1/CD133 — will contribute to a better understanding of SARS-CoV-2 cell entry, as outlined in the paper.
Best regards,
Pierre Brézellec.
References:
(Gabler et al., 2020):
Protein Sequence Analysis Using the MPI Bioinformatics Toolkit Felix Gabler, Seung-Zin Nam, Sebastian Till, Milot Mirdita, Martin Steinegger, Johannes Söding, Andrei N. Lupas, Vikram Alva. Current Protocols in Bioinformatics. December 2020.
(van Kempen & al., 2023):
Fast and accurate protein structure search with Foldseek. van Kempen M, Kim S, Tumescheit C, Mirdita M, Lee J, Gilchrist CLM, Söding J, and Steinegger M. Nature Biotechnology, 2023.
(Kelley & al., 2015):
The Phyre2 web portal for protein modeling, prediction and analysis. Lawrence A Kelley, Stefans Mezulis, Christopher M Yates, Mark N Wass & Michael J E Sternberg. Nature Protocols volume 10, pages 845–858. 2015.
(Söding, 2005):
Protein homology detection by HMM–HMM comparison. Johannes Söding. Bioinformatics, Volume 21, Issue 7, April 2005.
Decision by Jitendra Narayan, posted 22 Jun 2024, validated 02 Jul 2024
I have thoroughly examined the paper by Pierre Brézellec, titled "Re-annotation of SARS-CoV-2 proteins using an HHpred-based approach opens new opportunities for a better understanding of this virus." After carefully evaluating its content, methodology, and conclusions, I find the approach taken to seem overly simplistic for drawing meaningful conclusions. The author utilizes HHpred, a tool for protein homology detection and structure prediction, to identify similarities between SARS-CoV-2 and human proteins.
One of their primary claims is that the SARS-CoV-2 Spike protein is similar to the prominin domain of a human protein. However, this conclusion is based solely on profile-based searches, which compare sequence profiles rather than employing more robust structural or functional analyses. This approach may be considered somewhat crude and may not provide reliable or accurate insights into protein function or interactions (1,2,3). Additionally, the paper's second major conclusion involves the ORF3a protein of SARS-CoV-2. The authors themselves describe this finding as non-significant, raising further concerns about the robustness of their overall methodology and the validity of their conclusions **. Given these issues, I find the results presented in the paper to be questionable. The reliance on simplistic, profile-based methods and the acknowledgment of non-significant findings by the authors suggest that more rigorous approaches are needed to support the claims made in this study.
I recommend using multiple computational tools and databases for homology detection and structure prediction, such as PSI-BLAST and SWISS-MODEL, to cross-validate the findings from HHpred. Consistent results across different tools would enhance the validity of the conclusions. Additionally, conducting phylogenetic analysis to trace the evolutionary relationships between SARS-CoV-2 proteins and their human counterparts can provide further context on the functional and structural conservation of these proteins across different species. It is also advisable to use co-evolution analysis tools like GREMLIN or EVcouplings to identify co-evolving residues that might indicate functional interactions between SARS-CoV-2 proteins and human proteins.
** More precisely, none of the similarities found by HHpred are significant, i.e. the probability of the best hit is 0.72, which is below our threshold of 0.8; moreover, this best hit does not correspond to a GPCR (see Supplemental file 4). This result may suggest a lack of sensitivity of HHpred.
1. Jones, S., & Thornton, J. M. (1996), Principles of protein-protein interactions. Proceedings of the National Academy of Sciences, 93(1), 13-20. doi:10.1073/pnas.93.1.13).
2. Jeffery CJ (2023) Current successes and remaining challenges in protein function prediction. Front. Bioinform. 3:1222182. doi: 10.3389/fbinf.2023.1222182
3. Xiaoqing Peng, Jianxin Wang, Wei Peng, Fang-Xiang Wu, Yi Pan, Protein–protein interactions: detection, reliability assessment and applications, Briefings in Bioinformatics, Volume 18, Issue 5, September 2017, Pages 798–819, https://doi.org/10.1093/bib/bbw066
Reviewed by anonymous reviewer 1, 05 Apr 2024
I would like to thank Dr Brézellec for addressing my previous comments. I find this version of the manuscript improved and I recommend it for publication.
Evaluation round #1
DOI or URL of the preprint: https://doi.org/10.1101/2023.06.06.543855
Version of the preprint: 1
Author's Reply, 07 Mar 2024
Dear recommender,
Please find my replies to the comments and suggestions of the two anonymous referees of my paper entitled "Re-annotation of SARS-CoV-2 proteins using an HHpred-based approach opens new opportunities for a better understanding of this virus".
Best regards,
Pierre Brézellec.
//////////////////////////////////////////////////////////////////////////////////////////////////////////////////////////////
Dear anonymous referee 1,
Here are my replies to your relevant suggestions and comments.
#############################################################################################################################
1) First of all, I failed to understand whether when the author looked for hits, those viral proteins were already screened for "closer" homology. Do not all those proteins have other similarity apart from the human ones (e.g coronavirus or other virus proteins, bacterial, etc.)? If they do, how does this reconcile with the author's results?
2) Why and how did the author select human proteome only? I think it's because this proteome is the most characterized, but at the same time, could this create biases?
#############################################################################################################################
You address a very interesting and important point, so let me answer you after recalling the state of the art:
Accessory proteins of SARS-CoV-2 have been compared to accessory proteins from known coronaviruses (Forni et al., 2022). In
another work, each SARS-CoV-2 protein was modeled with related 3D structures in the PDB, i.e., structures determined for
other coronaviruses, such as SARS‐CoV or MERS‐CoV, as well as many structures from more distantly related viruses, such as
those causing polio or foot‐and‐mouth disease (O'Donoghue et al., 2021).
Moreover, as mentionned in section "Comparison of our results with those of "Pfam clans" of the paper", if among the 40 Pfam
domains that annotate SARS-CoV-2 proteins, only one domain is not confined to viruses, at the level of Pfam clans, 12 domains
belong to clans whose domains are not strictly viral (see Supplemental file 1). Thus, for instance, NSP16 - a
methyltransferase - is annotated with the Pfam clan NADP_Rossmann (CL0063), which annotates viruses, archaea, bacteria, and
eukaryotic proteins.
To conclude the state of art, it is established that the proteins of SARS-CoV-2 clearly resemble those of other viruses,
but not exclusively.
To date, to the best of my knowledge, nobody has published a work in which SARS-CoV-2 proteins (and more generally, proteins
from viruses that infect humans) are directly compared to human ones. This is quite strange, as it is now documented that
some viral proteins share similarities with their hosts.
From my point of view, there are two main reasons for this:
1./ Viral proteins that closely resemble host proteins are few in number.
2./ This has undoubtedly been attempted before, but without success.
What is new is the availability, provided by HHpred, of a database of Hidden Markov Models specific to Homo sapiens
proteins; what was previously achievable at the domain level now extends to human proteins.
I addressed your comments by adding and clarifying some sentences in the beginning of the paper.
Finally, note that in order to assess the relevance of the hits found by HHpred, I also used the following HH-suite custom
databases/proteomes: Arabidopsis thaliana, Drosophila melanogaster, Escherichia coli, or Haloferax volcanii.
#############################################################################################################################
2) Given the importance of this virus, most of its proteins are deeply analyzed. For example, it is well known that NSP16 is a methyltransferase, or some of the new findings reported here were already suggested. Maybe the author could add at the beginning of each paragraph some information for that specific protein.
#############################################################################################################################
You are correct: in section "A "highly robust" or "very robust" similarity, already documented in literature, was detected on
the following 4 proteins now briefly described", I give a brief description of each of the proteins.
#############################################################################################################################
"3) Following the previous point, a recent work showed that orf3a is not an ion channel (doi: 10.7554/eLife.84477). Maybe the author could discuss more about that. Regarding ORF3a, it seems too speculative to draw conclusions regarding GPCR similarity and autoimmunity."
#############################################################################################################################
I missed this article. Thank you for bringing it to my attention! I have cited and commented the findings of this paper in
the article.
As for the discussion regarding autoimmunity, you are completely right: it is all too speculative, and I have removed any
related content from the article.
#############################################################################################################################
4) the author stated that NSP13 has several human hits. Are those hits located in the same NSP13 region, or they spawn the whole protein length?
#############################################################################################################################
I believe you are experiencing a slight confusion. Actually, many human proteins share similarities with NSP13. However, all
these proteins are described with the same Pfam domains.
#############################################################################################################################
3) Hits found here are quite diverse in terms of aa length. Can protein length influence HHpred results?
#############################################################################################################################
Good question! Undoubtedly, it must have an impact; that being said, the length of the majority of the found similarities in
this work is quite large (e.g., approximately 350 AA for the similarity found on Spike S and 150 for the one found on ORF3a).
#############################################################################################################################
5) In the discussion section, line 385-395: I found this section more suitable for the result section.
#############################################################################################################################
Your viewpoint is valid. However, for a sake of clarity, I prefer to include in the results section only the previously undocumented similarities involving SARS-CoV-2 proteins. Note that, to comply with a suggestion made by the other referee,
the results section now includes only the two similarities (found in Spike S and ORF3a) not yet documented in the literature.
#############################################################################################################################
5) line 236 NSP13 should be NSP16.
#############################################################################################################################
You are correct. I have made the modification.
I thank you very much for your comments and suggestions.
Best regards,
Pierre Brézellec.
//////////////////////////////////////////////////////////////////////////////////////////////////////////////////////////////
//////////////////////////////////////////////////////////////////////////////////////////////////////////////////////////////
//////////////////////////////////////////////////////////////////////////////////////////////////////////////////////////////
Dear anonymous referee 2,
Thank you very much for your insightful comments. Here are my replies to your relevant comments and suggestions:
##############################################################################################################################
The author uses HHpred distant homology recognition server to push the analysis of SARS-CoV-2 proteins beyond that provided
by Pfam based HMM annotations. While commendable, this effort fails short of being novel or useful enough for publication.
The main reason is that with AlphaFold predicted 3D structures for all SARS-CoV-2 available or easily obtainable, much more
detailed analysis is possible.
Not to mention the fact that experimental structures are available for several of the "predictions" presented here, so why
not use them to verify the results presented here?
In addition, many of the specific observations made by the author are questionable and showcase limitations of very limited
approach taken by the author (HHpred analysis against the Pfam family database without considering 3D structures nor the
context of the potential hit).
##############################################################################################################################
By bringing up AlphaFold, you have pointed out an area that I have not explored. It is worth noting that at the time of
writing my paper (and this remains true today), there was no evidence in the literature suggesting that the structures
computed by AlphaFold allowed to improve the annotations of the proteins expressed by the SARS-CoV-2 virus.
That being said, as it is obviously always possible that nobody has actually attempted to use AlphaFold, your remark deserves
to be seriously considered.
In order to achieve this task, we used Foldseek (van Kempen M, Kim S, Tumescheit C, Mirdita M, Lee J, Gilchrist CLM, Söding
J, and Steinegger M. Fast and accurate protein structure search with Foldseek. Nature Biotechnology, 2023). Foldseek aligns
the structure of a query protein against a database by describing tertiary amino acid interactions within proteins as
sequences over a structural alphabet.
We thus ran Foldseek on the 3D structures of the two proteins of SARS-CoV-2 where we found similarities that have not yet
been reported in the literature, i.e., ORF3a and Spike S. This can be easily achieved via the UniProt entry of each of the 2
considered proteins. For the search we select AlphaFold/Swiss-Prot v4 and AlphaFold/Proteome v4 databases.
According to UniProt, 5 3D structures are available for ORF3a (https://www.uniprot.org/uniprotkb/P0DTC3/entry#structure). We
ran Foldseek on each of these 5 structures. No relevant results were found.
We then focused on Spike S. Not surprisingly, numerous 3D structures are available for Spike S (1470). We picked up two 3D
structures, namely 6vxx (Nature, 2020) and 6vsb (Cell, 2020). No hits were found.
[
6vxx: Walls, A.C., Park, Y.J., Tortorici, M.A., Wall, A., McGuire, A.T., Veesler, D. (2020) Structure, Function, and
Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 181, 281-292
6vsb: Wrapp, D., Wang, N., Corbett, K.S., Goldsmith, J.A., Hsieh, C.L., Abiona, O., Graham, B.S., McLellan, J.S. (2020) Cryo-
EM structure of the 2019-nCoV spike in the prefusion conformation. Science 367: 1260-1263
]
##############################################################################################################################
AlphaFold predictions incorporate HHpred results
##############################################################################################################################
In the Methods section of (Jumper et al., Nature 2021), it is indeed specified that "For MSA search on BFD + Uniclust30, and
template search against PDB70, we used HHBlits and HHSearch from hh-suite v.3.0-beta.3 (version 14/07/2017)".
However:
i/ AlphaFold uses the results generated by HHBlits and HHSearch to predict 3D structures, whereas HHpred uses these results
for a much more modest yet undoubtedly less risky task: generating a HMM for searching proteins "fitting the HMM".
ii/ Like HHpred, AlphaFold uses, among other databases, Uniclust30. However, it is not Uniclust30 that we use here but a
database centered on the proteome of Homo sapiens, i.e., Homo sapiens "custom HH-suite database" (https://github.com/
soedinglab/hh-suite/wiki#building-customized-databases). This custom HH-suite database is based on Uniclust30, but it likely
possesses different properties. We emphasize this point in the paper:
"We speculated that this difference might give HHpred the ability to discover similarities not detectable by Pfam (it should
be noted that a theoretical comparison between the Pfam and HHpred HMMs, as well as a full empirical comparison, is beyond
the scope of this paper)"
In conclusion, in our work, the results we use are not exactly those of AlphaFold; furthermore, and this is the most
important point, these results are used to achieve very different purposes!
##############################################################################################################################
"Because of specific sequence signatures of transmembrane helical proteins they are notoroius for false positive (at least in the functional sense) predictions. Similar pattern of a transmembrane helix - exposed loop - TM helix etc. can be seen in many unrelated proteins with disparate functions."
##############################################################################################################################
This is correct and part "Evaluation of our results in light of the known weaknesses of HHpred" of the
paper addresses the valid criticisms you mentioned.
##############################################################################################################################
"as can be seen in the experimental structure of nsp2 protein, the 151-195 fragment does not form a domain, but is spread between two mini-domains".
##############################################################################################################################
You are correct. The method we use does not rely on the notion of domain. We clarify this point in the new version of the
paper.
##############################################################################################################################
"- most other results (NSP3 Macro domain, NSP16 as a methyltransferase) were very well known in literature, perhaps not picked up by Pfam, but shouldnt be presented as "results"
##############################################################################################################################
You are correct. We have changed this way of presenting things in the new version of the article.
##############################################################################################################################
"the suposedy AAA structure in nsp13 should be compared to the known experimental structure of nsp13"
##############################################################################################################################
NSP13 is a helicase, and more precisely a RNA virus helicase (https://www.ebi.ac.uk/interpro/entry/InterPro/IPR027351/). It
belongs to the "P-loop containing nucleoside triphosphate hydrolase (IPR027417)" superfamily
which AAA domains are members (https://www.ebi.ac.uk/interpro/entry/InterPro/IPR027417/).
To conclude:
1./ I cannot include the results obtained with Foldseek/AlphaFold as as they do not provide any additional value to the
annotations.
2./ However, I have taken into account your suggestions regarding what should belong to the results section and what clearly
should not. This increases the clarity of the paper. I thank you very much.
3./ I acknowledge that the results obtained in my paper are modest. However, even modest progress can sometimes lead to
interesting discoveries and/or stimulating hypotheses, in particular, when the annotation improvement concerns the Spike S
protein, what is the case here.
Thank you,
Best regards,
Pierre Brézellec.
//////////////////////////////////////////////////////////////////////////////////////////////////////////////////////////////
Decision by Jitendra Narayan, posted 17 Jan 2024, validated 17 Jan 2024
The reviewers offered constructive criticisms and provided insightful comments - second review raises potentially major issues that question the relevance of the work, which the author will definitely need to clearly respond to. Please revise your manuscript accordingly and provide a point-by-point reply to the reviews outlining the changes you made and elaborating on any additional data or analyses you performed. I look forward to reading the next version of your preprint.
Reviewed by anonymous reviewer 1, 30 Oct 2023
In this article, the author uses an in silico approach to characterize SARS-CoV-2 proteins/protein domains. This is an interesting approach, particularly as it is based on distant homologies and, as the author states, the molecular mimicry of viral protein is a field with an increasing interest.
I only have a few comments and suggestions to the author.
1) First of all, I failed to understand whether when the author looked for hits, those viral proteins were already screened for "closer" homology. Do not all those proteins have other similarity apart from the human ones (e.g coronavirus or other virus proteins, bacterial, etc.)? If they do, how does this reconcile with the author's results?
2) Given the importance of this virus, most of its proteins are deeply analyzed. For example, it is well known that NSP16 is a methyltransferase, or some of the new findings reported here were already suggested. Maybe the author could add at the beginning of each paragraph some information for that specific protein.
3) Following the previous point, a recent work showed that orf3a is not an ion channel (doi: 10.7554/eLife.84477). Maybe the author could discuss more about that. Regarding ORF3a, it seems too speculative to draw conclusions regarding GPCR similarity and autoimmunity.
2) Why and how did the author select human proteome only? I think it's because this proteome is the most characterized, but at the same time, could this create biases?
3) Hits found here are quite diverse in terms of aa length. Can protein length influence HHpred results?
4) the author stated that NSP13 has several human hits. Are those hits located in the same NSP13 region, or they spawn the whole protein length?
5) In the discussion section, line 385-395: I found this section more suitable for the result section.
5) line 236 NSP13 should be NSP16.
Reviewed by anonymous reviewer 2, 27 Nov 2023
The author uses HHpred distant homology recognition server to push the analysis of SARS-CoV-2 proteins beyond that provided by Pfam based HMM annotations. While commendable, this effort fails short of being novel or useful enough for publication. The main reason is that with AlphaFold predicted 3D structures for all SARS-CoV-2 available or easily obtainable, much more detailed analysis is possible. BTW, AlphaFold predictions incorporate HHpred results, so it can can add and further clarify the HHpred analysis and should not be ignored, especially >2 years since their availability. Not to mention the fact that experimental structures are available for several of the "predictions" presented here, so why not use them to verify the results presented here?
In addition, many of the specific observations made by the author are questionable and showcase limitations of very limited approach taken by the author (HHpred analysis against the Pfam family database without considering 3D structures nor the context of the potential hit). For instance:
- hits of orf3a and the section of the spike protein are formed by transmembrane helices. Because of specific sequence signatures of transmembrane helical proteins they are notoroius for false positive (at least in the functional sense) predictions. Similar pattern of a transmembrane helix - exposed loop - TM helix etc. can be seen in many unrelated proteins with disparate functions.
- as can be seen in the experimental structure of nsp2 protein, the 151-195 fragment does not form a domain, but is spread between two mini-domains.
- most other results (NSP3 Macro domain, NSP16 as a methyltransferase) were very well known in literature, perhaps not picked up by Pfam, but shouldnt be presented as "results"
- the suposedy AAA structure in nsp13 should be compared to the known experimental structure of nsp13.




