
What does dental gene decay tell us about the regressive evolution of teeth in South American mammals?
Genomic data suggest parallel dental vestigialization within the xenarthran radiation
Abstract
Recommendation: posted 03 August 2023, validated 07 August 2023
Casane, D. (2023) What does dental gene decay tell us about the regressive evolution of teeth in South American mammals?. Peer Community in Genomics, 100240. 10.24072/pci.genomics.100240
Recommendation
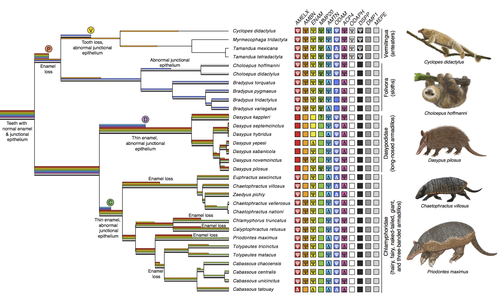
A group of mammals, Xenathra, evolved and diversified in South America during its long period of isolation in the early to mid Cenozoic era. More recently, as a result of the Great Faunal Interchange between South America and North America, many xenarthran species went extinct. The thirty-one extant species belong to three groups: armadillos, sloths and anteaters. They share dental degeneration. However, the level of degeneration is variable. Anteaters entirely lack teeth, sloths have intermediately regressed teeth and most armadillos have a toothless premaxilla, as well as peg-like, single-rooted teeth that lack enamel in adult animals (Vizcaíno 2009). This diversity raises a number of questions about the evolution of dentition in these mammals. Unfortunately, the fossil record is too poor to provide refined information on the different stages of regressive evolution in these clades. In such cases, the identification of loss-of-function mutations and/or relaxed selection in genes related to a character regression can be very informative (Emerling and Springer 2014; Meredith et al. 2014; Policarpo et al. 2021). Indeed, shared and unique pseudogenes/relaxed selection can tell us to what extent regression has occurred in common ancestors and whether some changes are lineage-specific. In addition, the distribution of pseudogenes/relaxed selection on the branches of a phylogenetic tree is related to the evolutionary processes involved. A much higher density of pseudogenes in the most internal branches indicates that degeneration took place early and over a short period of time, consistent with selection against the presence of the morphological character with which they are associated, while pseudogenes distributed evenly in many internal and external branches suggest a more gradual process over many millions of years, in line with relaxed selection and fixation of loss-of-function mutations by genetic drift.
In this paper (Emerling et al. 2023), the authors examined the dynamics of decay of 11 dental genes that may parallel teeth regression. The analyses of the data reported in this paper clearly point to xenarthran teeth having repeatedly regressed in parallel in the three clades. In fact, no loss-of-function mutation is shared by all species examined. However, more genes should be studied to confirm the hypothesis that the common ancestor of extant xenarthrans had normal dentition. There are distinct patterns of gene loss in different lineages that are associated with the variation in dentition observed across the clades. These patterns of gene loss suggest that regressive evolution took place both gradually and in relatively rapid, discrete phases during the diversification of xenarthrans. This study underscores the utility of using pseudogenes to reconstruct evolutionary history of morphological characters when fossils are sparse.
References
Emerling CA, Gibb GC, Tilak M-K, Hughes JJ, Kuch M, Duggan AT, Poinar HN, Nachman MW, Delsuc F. 2023. Genomic data suggest parallel dental vestigialization within the xenarthran radiation. bioRxiv, 2022.12.09.519446, ver 2, peer-reviewed and recommended by PCI Genomics. https://doi.org/10.1101/2022.12.09.519446
Emerling CA, Springer MS. 2014. Eyes underground: Regression of visual protein networks in subterranean mammals. Molecular Phylogenetics and Evolution 78: 260-270. https://doi.org/10.1016/j.ympev.2014.05.016
Meredith RW, Zhang G, Gilbert MTP, Jarvis ED, Springer MS. 2014. Evidence for a single loss of mineralized teeth in the common avian ancestor. Science 346: 1254390. https://doi.org/10.1126/science.1254390
Policarpo M, Fumey J, Lafargeas P, Naquin D, Thermes C, Naville M, Dechaud C, Volff J-N, Cabau C, Klopp C, et al. 2021. Contrasting gene decay in subterranean vertebrates: insights from cavefishes and fossorial mammals. Molecular Biology and Evolution 38: 589-605. https://doi.org/10.1093/molbev/msaa249
Vizcaíno SF. 2009. The teeth of the “toothless”: novelties and key innovations in the evolution of xenarthrans (Mammalia, Xenarthra). Paleobiology 35: 343-366. https://doi.org/10.1666/0094-8373-35.3.343

The recommender in charge of the evaluation of the article and the reviewers declared that they have no conflict of interest (as defined in the code of conduct of PCI) with the authors or with the content of the article. The authors declared that they comply with the PCI rule of having no financial conflicts of interest in relation to the content of the article.
This research was supported by a European Research Council consolidator grant (ConvergeAnt ERC-2015-CoG-683257; FD); the Centre National de la Recherche Scientifique (CNRS; FD); the Scientific Council of the Université de Montpellier (FD); Investissements d’Avenir grants managed by Agence Nationale de la Recherche (CEBA: ANR-10-LABX-25-01; CEMEB: ANR-10-LABX-0004; FD); a National Science Foundation Postdoctoral Research Fellowship in Biology (award no. 1523943; CAE); a National Science Foundation Postdoctoral Fellow Research Opportunities in Europe award (CAE); the People Programme (Marie Curie Actions) of the European Union’s Seventh Framework Programme (FP7/2007-2013) under REA grant agreement no. PCOFUND-GA-2013-609102, through the PRESTIGE programme coordinated by Campus France (CAE); the France-Berkeley Fund (FD and MWN); and the Natural Sciences and Engineering Research Council of Canada (NSERC, no. RGPIN04184-15) and the Canada Research Chairs program (HNP).
Evaluation round #1
DOI or URL of the preprint: https://doi.org/10.1101/2022.12.09.519446
Version of the preprint: 1
Author's Reply, 01 Aug 2023
Dear Editors,
We are pleased to submit a revised version of our preprint entitled “Genomic data suggest parallel dental vestigialization within the xenarthran radiation” for potential recommendation by Peer Community in Genomics.
We would like to thank the recommender and the three referees for their helpful comments that we have taken into account. Our point-by-point answers (in blue) are detailed in the attached pdf file.
With this improved revised version, we hope that our manuscript now meets the standards for recommendation by Peer Community in Genomics.
Yours sincerely,
Frédéric Delsuc on the behalf of all co-authors.
Decision by Didier Casane, posted 31 Jan 2023, validated 01 Feb 2023
Decision: revise
Three reviewers gave very positive feedback on this manuscript. They also suggest that some changes be made to improve its readability and understanding of the results by most readers. I suggest that the most important ones be done. Many minor concerns should also be considered too.
1) The authors assembled a dataset of 11 dental genes: nine genes having well-characterized functions and/or expression patterns tied to tooth development and frequently pseudogenized in edentulous and enamelless taxa, and two other genes expressed in teeth but do not have the same patterns of inactivation. I understand this selection that makes sense, but I was wondering if it could be possible to enlarge the set of genes, for example by looking for pseudogenes in xenarthran genomes and checking those involved in teeth development using mammal gene expression databases. I was wondering if these 11 genes represent a small or a large subset of teeth specific genes. If it is a small subsets and if the ancestral branch is small compared to the xenarthran tree, even if some pseudogenes are shared by all xenarthrans, because there are few of them we need a large gene set to find those that appeared in the common ancestor. Thus, no common loss of function mutations in tooth specific genes could be the consequence of a too small number of genes examined and not evidence of normal teeth and gingiva in the common ancestor.
2) A reviewer wrote: Regarding the methodology, although I understand that they accomplish the goal of getting the sequence of the genes using different approaches, adding a schematic figure showing which method was used for which species would help to understand more easily. Another reviewer wrote: this data collection might also be one of the few limitations of the paper: even with such a broad effort, a lot of genetic information is missing from the final dataset. Except for the few fully sequenced analyzed, it is unclear whether the missing exons of various genes are the reflection of a biological reality or rather of the incomplete molecular sampling due to imperfect amplification or capture and/or lack of depth in the sequencing. This difficulty is exemplified by the DMP1 and MEPE datasets (see below). Since these missing pieces of genes might themselves be involved in the identification of inactivated genes, it slightly blurs the general message. The limited completeness of the dataset for this marker (and the others too) should be directly accessible from the main text to be obvious to help contextualize the interpretation.
Thus, for the 11 genes examined, a clear description of the data completeness is necessary.
3) A reviewer wrote: I also like the evolutionary rationale behind reconstructing dN/dS values and when the inactivation process occurred in the xenarthran phylogeny. I recommend creating a schematic figure showing the rationale. Being more didactic will open the paper to a broader audience.
I agree, although crafting such a figure may not be an easy task.
Reviewed by Juan C. Opazo , 30 Dec 2022
, 30 Dec 2022
Download the reviewReviewed by Régis Debruyne, 17 Jan 2023
The manuscript entitled “Genomic data suggest parallel dental vestigialization within the xenarthran radiation” by C.A. Emerling et al. presents evidence for the parallel evolution and gradual decay in dental regression in xenarthran lineages. It is a very consistent and robust manuscript, based on a well-designed analytical methodology.
The authors ground their hypotheses on two major lines of evidence: sequence analysis of gene pseudogenization and dN:dS analysis of nucleotide sequences, for eleven core genes involved in enamel and tooth formation. Both provide compelling support to the main hypotheses of independent and stepwise loss for enamel/teeth.
The scientific background and the specific aims of the manuscript are well-developed while remaining clear to the non-specialist. The manuscript is well-written and the authors provide a wealth of details about how the dataset was constructed and analyzed. The in-text figures are designed clearly while conveying a large amount of information. All necessary material is presented or made available to support the authors’ claims; relevant supplementary tables (26 six of them, all meaningful to me) and figures also provide extra details to the keen reader. I appreciated that the discussion addresses how the molecular findings integrate with the (scarce) paleontological evidence available from the relevant Eocene/Oligocene Xenarthra fossils, and the changes in the ecology/diet of the sloths.
I only regret that some sentences in the main text sometimes lean towards a non-necessary finalist presentation of the evolutionary processes. For example l.34 reads “sloths halted their dental regression; l.468-9 reads “stem dasypodid armadillos independently inactivated AMTN”. My remark might only reflect the fact that English is not my mother tongue, and that this phrasing sounds finalist to the French reader that I am.
I am fully convinced by the various claims made in the manuscript concerning the independence, parallelism, and gradual characteristics of the dental regression processes identified in the various subclades within Xenarthra. I agree with the authors that the lack of SIM signal observed in the DMP1 and MEPE genes only reinforces the observations made on the 9 other genes more specifically involved in enamel/tooth development.
I am a bit surprised that not a single SIM was recovered for any of the genes under scrutiny for the ancestral branch of the entire Xenarthra super-order, considering that dental regression is a feature observed even in the earliest-known xenarthran fossils (as indeed the authors acknowledge in the discussion). I think that this observation might deserve a little more consideration in the discussion: it could be emphasized that this is a clear limitation of such a functional study based solely on the analysis of coding sequences and that the processes identified here might just reflect one side of the coin. Adding to this, I wonder why only the coding sequences of the focal genes were targeted. It is obvious from the results (AMTN, ODAM) that key mutations leading to pseudogenization might have occurred in non-coding sequences of these genes or promoter regions also. Were the intron sequences excluded for any specific reason (like strong divergence among xenarthrans, or else)? I would appreciate the authors briefly justify this choice.
Gathering the gene (exonic) sequences dataset analyzed in the paper for such an extensive sample within modern Xenarthrans (31 species represented) is quite impressive. The sources of the data analyzed are extremely diverse: whole-genome sequencing, shotgun and target capture sequencing, targeted amplicon sequencing. Yet, this data collection might also be one of the few limitations of the paper: even with such a broad effort, a lot of genetic information is missing from the final dataset. Except for the few fully sequenced analyzed, it is unclear whether the missing exons of various genes are the reflection of a biological reality or rather of the incomplete molecular sampling due to imperfect amplification or capture and/or lack of depth in the sequencing. This difficulty is exemplified by the DMP1 and MEPE datasets (see below). Since these missing pieces of genes might themselves be involved in the identification of inactivated genes, it slightly blurs the general message.
For the ACPT gene, specifically, the authors state that its CDSs were not included in the baits designed for target sequence. Why? Due to this choice, a majority (19 out of 31) of xenarthran taxa are not directly represented in the ACPT alignment. Thus direct evidence of pseudogenization for this gene is scarce. However, figure 1 indicates that for 11 non-sequenced armadillo species, the dN/dS ratio estimates suggest a possible gene inactivation. Could the authors be more specific as to how this result is obtained since only a single direct sequence is available for all 12 most basal armadillos?
When compared with the information conveyed in figure 1, it seems that some of the identifications for the genes between ‘missing-or-pseudogene_phylogeny-based/delta’, ‘pseudogenized/psi’, ‘unknown/?’ and ‘pseudogene_dN:dS-based/omega’ lack consistency among the various markers and taxa. For example, some ‘delta’ identifications are made for some taxa for which not a single exon sequence of the marker of interest was retrieved (as ‘missing’), but sometimes it is given for taxa that show only a few of the marker exons (as pseudogene based on the phylogeny’). I would rather have these two situations split into two different summary letters, for they do not convey the same information at all.
I’ll provide a few examples, below, of situations that put the emphasis on the not-so-obvious link between the supplementary tables S5 to S15 and the interpretation that is made of them within figure 1.
Table S6 – AMBN gene
The sequence for Cabassous tatouay is represented in the dataset by a single exon (exon6) which is deemed putatively functional, but the other 9 expected exons are missing. This sequence shows a “?” in figure 1. However, nine other taxa lack one or several exons (up to 5 for Euphractus sexcinctus) for this gene and yet are all deemed functional for AMBN, because no obvious signature of pseudogenization is found in the remaining exons.
Isn’t there a risk, in such a situation – which corresponds to the majority of the markers analyzed – to underestimate the actual inactivation of the markers? In other words, how can we be confident that, with one or several missing exons, the protein produced is still efficient and not sub-functional? And from how many absent exons should one deem that the protein is not functional anymore? It seems fairly arbitrary to me at this stage (and if so, which is fine, it should at least be specified).
Table S7 – AMELX gene
For Cabassous tatouay, again, only one exon out of 4 is present in the dataset (and not mutated). Yet this time the gene is deemed inactivated via ‘delta’ in figure 1. Why is the interpretation different for this marker from the AMBN case?
Table S8 – AMTN gene
Priodontes maximus shows only one (non-mutated) exon out of the 7 expected and is identified with a delta (pseudogenized based on phylogeny I guess) like Totypeutes tricinctus for which not a single exon was found (thus recorded as a missing gene for this one). Again the use of a delta for both categories seems misleading to me.
Table S14 – ODAM mutations
Among Vermilingua, Myrmecophaga tridactyla show the 10 expected exons of the marker, with 6 of them showing inactivating mutations, and yet it is identified as a “delta” - inactivated based on the phylogenetic bracketing – instead of the “psi” like Tamandua tetradactyla. Why?
Table S9 – DMP1
All 31 taxa are presented as having a functional DMP1 in figure 1. Yet, the 5 expected exons are only documented for the 11 complete genomes analyzed, whereas the other 20 taxa showed the lack of at least 1 and up to 4 of these exons. Based on the observation that the present exons never show a SIM signature, it sounds reasonable to extrapolate that these sequences are functional as the authors do. Yet, the limited completeness of the dataset for this marker (and the others too) should be directly accessible from the main text to be obvious to help contextualize the interpretation made.
Reviewed by Nicolas Pollet, 30 Dec 2022
Dear Christopher Emerling and colleagues,
I read with interest your preprint entitled « Genomic data suggest parallel dental vestigialization within the xenarthran radiation ». In this research article, you report your findings on the patterns of sequence evolution related to the vestigialization of teeth development in xenarthran, a group of mammals known to have experienced changes in their teeth ranging from regressive modifications to a complete absence.
You collected data and explored the evolution of sequences corresponding to eleven known genes involved in tooth development in anteaters, armadillos and slugs. You present a phylogenomic analysis that revealed different patterns of mutations across Xenarthra. You conclude that the regressive evolution linked to tooth development in these mammals could not be linked to a single mutation, and instead followed parallel trajectories in the different clades and occurred relatively rapidly.
I find that the title reflects the main message of the study.
I find that the abstract is relatively concise and presents the main results and conclusions of the study.
In the introduction, the theme of “regressive” evolution is presented as the underlying theme of the paper. The specific domain of teeth loss in jawed vertebrates is also explained, along with background information on specific and well-known genes involved in tooth development and differentiation. The Xenarthra taxon and its teeth traits are presented in enough detail so that the reader can understand the research questions and hypothesis. The references provided in the introduction are relevant and cover not only the most recent research but also covers older literature.
In the Materials and methods, the information related to the gene set being analysed is presented in sufficient details. I just checked the gene symbols and became aware that the approved gene symbol for ACPT is now ACP4 (and at the same time I learned that its whole name is testicular acid phosphatase ! https://www.genenames.org/data/gene-symbol-report/#!/hgnc_id/HGNC:14376). The taxonomic sampling is presented with details on each sample. Protocols for the experiments of molecular genetics are given in a manner enabling reproducibility. The sequences OP966064-OP966335 were not unavailable at the time of review. The procedure for sequence analysis are missing some minor details (e.g. BLAST version). The Bioproject PRJNA907496 was unavailable at the time of review. I have to say that I releasing the southern naked-tailed armadillo genome assembly as a Zenodo dataset is not acceptable, the international nucleotide sequence database collection (INSDC www.insdc.org) lists the sole repositories for such data.
In the paragraph Dataset Assembly, line 225 Supplementary dataset S1 does not correspond to sequence alignments, I guess you meant dataset S2-S23. In this paragraph, I was expecting to find the rationale to consider an exon as missing (ie yellow in supp tables S5-S15).
Line 219 you mention that “sequences were assembled and occasionally combined”, how does this link to the supp table 2 ? Did you use a cut-off for base quality before proceeding to mutation analysis and did you look for hetero or homozygosity in the case of inactivating mutations?
Overall there is no specific code or script provided. This latter point could be improved as the analysis of the branch model dN/dS ratio with the various models and their statistical analysis (lines 245-266) is of methodological interest.
In the results, you reported the findings on gene mutation patterns across xenarthrans in a condensed format in Figure 1. While this figure is extremely informative, I think that it holds too many pieces of information. I have a suggestion to help the reader follow the figure along with the text: you could position the matrix of shared inactivation on the left as it is the starting point in terms of data, and then have the species tree (with the root oriented toward the right). In my opinion, the mix of colors and symbols is extremely difficult to analyze. A possibility would be to split the figure in two, with one panel showing the data matrix, and another panel with the tree and traits. Also, the use of serif characters in figures is usually avoided (you used it for sub-order names and common names).
Line 410-414: I suggest that you follow the scientific convention on significant figures for omega values.
In Figure 2, which provides key data, I notice that the alignments of the different genes do not always include the translation, you could maybe insert a consensus translation over the alignments in these cases. The presentation of the different genes does not follow a simple rule (such as alphabetic ordering), so different mutations of the same gene are not presented side by side. Since the objective here is to show shared inactive mutations, It may be better to include all taxa for a given gene with different SIMs : e.g. AMTN exon1 next to exon4, with all taxa. And to highlight mutations, the use of a dot for identical residues and of lowercase for silent mutations may help.
On figure 3, the color scale is too small to be readable. In the text, you mention lines 423-427 that patterns of relaxed selection are strikingly similar among genes having the same or similar role during tooth development. This statement lacks some explicit description, as a matter of fact, I found that DSPP and MMP20 share a similar pattern, and that EDMA and ODAM also share a similar pattern. You could expand this section to draw conclusions that are more in line with the results.
Discussion:
Line 442 :I was wondering about the odds to identify shared inactivation mutations across the xenarthrans radiation estimated at 68 million years ago? Intuitively and maybe naively, I would expect that a single ancestral inactivating mutation could be followed by subsequent mutations of different scopes, including larger deletions. The finding that exon 11 of ACPT is deleted in most xenarthran would fit such a scenario.
Line 458 a typo at coeval ?
In this discussion, I was expecting you to provide some more elements on the limitations of your work and your approach to finding mutations.
Overall your interpretation of the data takes into account the older and newer studies in the field, and they are reasonable given the provided results. I especially appreciated the broader implications regarding gene loss and evolution, their tempo, and the relationship with life traits.
Supplementary data:
I was surprised to find the legends for the supplementary datasets and figures in the supplementary_Tables_S1-S26. I suggest you to add a text file for all supplementary datasets and figures caption, but since it is on the Zenodo page this may be unessential. Dataset S1 does not contain all FASTA alignments, I guess you omitted the range: Dataset S1-S24 !
The main strength of your study is that I find it extremely well performed, and associated with relevant conclusions.
The main weakness is that I could not find in the manuscript the details to ensure that all necessary permits or approvals were obtained for collecting the animals. I acknowledge that this can be difficult for Museum samples, but such details should be available for samples from the Animal Tissue Collection of ISEM, for which you gave voucher information. For these vouchers, I could no find any information on the permits that enabled animal collection, even from the cited references. The reference Gibb et al 2016 mention only two permits for Bradypus torquatus and Tolypeutes tricinctus. Moreover, some samples presented here are not described in Gibb et al 2016 (e.g T-1476, T-1722, T-1631, T-2977, T-JL556). Delsuc et al 2018 refers only to Mylodon darwinii. Whether we like it or not, this is an ethical requirement for a scientific publication. In my quality of reviewer, I should also point out that a Benefit-sharing statement is a condition for publication in numerous journals, such as Molecular Ecology (https://onlinelibrary.wiley.com/page/journal/1365294x/homepage/forauthors.html).




