
Recommendation
One way to identify all organisms at their various life stages is by their genetic signature. DNA-based taxonomy, gene tagging and barcoding are different shortcuts used to name such strategies (Lamb et al. 2019; Tautz et al. 2003). Reading and analyzing nucleic acid sequences to perform genetic inventories is now faster than ever, and the latest nucleic acid sequencing technologies reveal an impressive taxonomic, genetic, and functional diversity hidden in all ecosystems (Lamb et al. 2019; Sunagawa et al. 2015). This knowledge should enable us to evaluate biodiversity across its scales, from genetic to species to ecosystem and is sometimes referred to with the neologism of ecogenomics (Dicke et al. 2004).
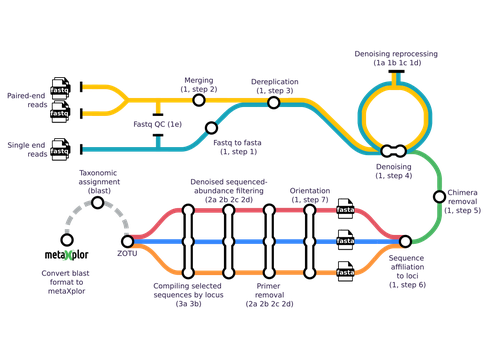
The metabarcoding approach is a key workhorse of ecogenomics. At the core of metabarcoding strategies lies the sequencing of amplicons obtained from so-called multi-template PCR, a formidable and potent experiment with the potential to unravel hidden biosphere components from different samples obtained from organisms or the environment (Kalle et al. 2014; Rodríguez-Ezpeleta et al. 2021). Next to this core approach, and equally important, lies the bioinformatic analysis to convert the raw sequencing data into amplicon sequence variants or operational taxonomic units and interpretable abundance tables.
Methodologically, the analysis of sequences obtained from metabarcoding projects is replete with devilish details. This is why different pipelines and tools have been developed, starting with mothur (Schloss et al. 2009) and QIIME 2 (Bolyen et al. 2019), but including more user friendly tools such as FROGS (Escudié et al. 2018). Yet, across all available tools, scientists must choose the optimal algorithms and parameter values to filter raw reads, trim primers, identify chimeras and cluster reads into operational taxonomic units. In addition, the number of genetic markers used to characterize a sample using metabarcoding has increased as sequencing methods are now less costly and more efficient. In such cases, results and interpretations may become limited or confounded. This is where the novel tools proposed by Barnabé and colleagues (2024), mbctools, will benefit researchers in this field.
The authors provide a detailed description with a walk-through of the mbctools pipeline to analyse raw reads obtained in a metabarcoding project. The mbctools pipeline can be installed under different computing environments, requires only VSEARCH and a few Python dependencies, and is easy to use with a menu-driven interface. Users need to prepare their data following simple rules, providing single or paired-end reads, primer and target database sequences. An interesting feature of mbctools output is the possibility of integration with the metaXplor visualization tool developed by the authors (Sempéré et al. 2021). As it stands, mbctools should be used for short-read sequences. The taxonomy assignment module has the advantage to enable parameters exploration in an easy way, but it may be oversimplistic for specific taxa.
The lightweight aspect of mbctools and its overall simplicity are appealing. These features will make it a useful pipeline for training workshops and to help disseminate the use of metabarcoding. It also holds the potential for further improvement, by the developers or by others. In the end, mbctools will support study reproducibility by enabling a streamlined analysis of raw reads, and like many useful tools, only time will tell whether it is widely adopted.
References
Barnabé C, Sempéré G, Manzanilla V, Millan JM, Amblard-Rambert A, Waleckx E (2024) mbctools: A user-friendly metabarcoding and cross-platform pipeline for analyzing multiple amplicon sequencing data across a large diversity of organisms. bioRxiv, ver. 2 peer-reviewed and recommended by PCI Genomics https://doi.org/10.1101/2024.02.08.579441
Bolyen E, Rideout JR, Dillon MR, Bokulich NA, et al. (2019) Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nature Biotechnology, 37, 852–857. https://doi.org/10.1038/s41587-019-0209-9
Dicke M, van Loon JJA, de Jong PW (2004) Ecogenomics benefits community ecology. Science, 305, 618–619. https://doi.org/10.1126/science.1101788
Escudié F, Auer L, Bernard M, Mariadassou M, Cauquil L, Vidal K, Maman S, Hernandez-Raquet G, Combes S, Pascal G (2018) FROGS: Find, Rapidly, OTUs with Galaxy Solution. Bioinformatics, 34, 1287-1294. https://doi.org/10.1093/bioinformatics/btx791
Kalle E, Kubista M, Rensing C (2014) Multi-template polymerase chain reaction. Biomolecular Detection and Quantification, 2, 11–29. https://doi.org/10.1016/j.bdq.2014.11.002
Lamb CT, Ford AT, Proctor MF, Royle JA, Mowat G, Boutin S (2019) Genetic tagging in the Anthropocene: scaling ecology from alleles to ecosystems. Ecological Applications, 29, e01876. https://doi.org/10.1002/eap.1876
Rodríguez-Ezpeleta N, Zinger L, Kinziger A, Bik HM, Bonin A, Coissac E, Emerson BC, Lopes CM, Pelletier TA, Taberlet P, Narum S (2021) Biodiversity monitoring using environmental DNA. Molecular Ecology Resources, 21, 1405–1409. https://doi.org/10.1111/1755-0998.13399
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Applied and Environmental Microbiology 75, 7537-41. https://doi.org/10.1128/AEM.01541-09
Sempéré G, Pétel A, Abbé M, Lefeuvre P, Roumagnac P, Mahé F, Baurens G, Filloux D 2021 metaXplor: an interactive viral and microbial metagenomic data manager. Gigascience, 10, https://doi.org/10.1093/gigascience/giab001
Sunagawa S, Coelho LP, Chaffron S, Kultima JR, Labadie K, Salazar G, Djahanschiri B, Zeller G, Mende DR, Alberti A, Cornejo-Castillo FM, Costea PI, Cruaud C, d’Ovidio F, Engelen S, Ferrera I, Gasol JM, Guidi L, Hildebrand F, Kokoszka F, Lepoivre C, Lima-Mendez G, Poulain J, Poulos BT, Royo-Llonch M, Sarmento H, Vieira-Silva S, Dimier C, Picheral M, Searson S, Kandels-Lewis S, Tara Oceans coordinators, Bowler C, de Vargas C, Gorsky G, Grimsley N, Hingamp P, Iudicone D, Jaillon O, Not F, Ogata H, Pesant S, Speich S, Stemmann L, Sullivan MB, Weissenbach J, Wincker P, Karsenti E, Raes J, Acinas SG, Bork P (2015) Structure and function of the global ocean microbiome. Science, 348, 1261359. https://doi.org/10.1126/science.1261359
Tautz D, Arctander P, Minelli A, Thomas RH, Vogler AP (2003) A plea for DNA taxonomy. Trends in Ecology & Evolution, 18, 70–74. https://doi.org/10.1016/S0169-5347(02)00041-1
DOI or URL of the preprint: https://doi.org/10.1101/2024.02.08.579441
Version of the preprint: 2
Dear Christian Barnabé and co-authors,
I looked through both reviews and I agree with the reviewers that your manuscript presents a valuable tool. Provided minor revisions along the lines given by the reviewers, you could easily improve the quality of your communication.
 , 02 Apr 2024
, 02 Apr 2024The manuscript presents a valuable tool for the metabarcoding community, effectively addressing a gap in the availability of accessible, flexible, and comprehensive analysis software. With minor revisions, particularly aimed at enhancing clarity, user guidance, and the provision of additional context and examples, the manuscript would be a strong candidate for publication. The suggested revisions are not expected to require substantial changes to the content or structure of the manuscript but rather to enrich the existing sections with additional details and clarifications.
Abstract
The abstract provides a clear and concise overview of the mbctools package, its functionalities, and its advantages. However, there are several areas where clarity and detail could be enhanced:
Line 21–27: It is commendable that the tool is designed for users without command-line expertise, but it would be beneficial to briefly mention the specific user interface design or examples of how the menu-driven program simplifies the process.
Line 28–32: While VSEARCH's utilization is noted, a brief explanation of why VSEARCH was chosen over other tools, considering its advantages in processing amplicon data, would provide a more comprehensive background.
Introduction
The introduction effectively sets the stage by highlighting the importance and applications of eDNA metabarcoding. It also identifies a gap in the availability of user-friendly bioinformatics tools. Nevertheless, some aspects could be refined for improved clarity and engagement:
Lines 39–52: The introduction to eDNA metabarcoding and its applications is well-articulated. However, referencing specific studies (1–3, 4, 5, 6–8, 9, 10) without any context may leave readers unfamiliar with the cited works without a clear understanding. Brief descriptions of these references could enhance the introduction's informativeness.
Lines 53–64: This section does well in identifying a gap in current bioinformatics tools. Briefly addressing the specific difficulties that novice users have using current tools would strengthen it and make the case for the development of mbctools more compelling.
Lines 65–78: The rationale for choosing VSEARCH and the description of mbctools' functionality are clear. However, stating that "the software can be used only with command lines" (Line 77) seems to contradict the abstract's emphasis on a user-friendly, menu-driven interface. Clarification on this point would be helpful. Additionally, illustrating how mbctools compares to or improves upon existing tools in sensitivity, specificity, or user-friendliness would add valuable context for the reader.
Figures1-2-3: The quality of the figures appears to be quite low; I recommend using photos with greater resolution and better display. They appear to be not original creations, and designing in applications like Canva, POWERPOINT, or Inkscape can yield superior outcomes.
Prerequisites and Dependencies
The "Prerequisites and Dependencies" section provides a detailed overview of the technical requirements and setup needed to utilize mbctools efficiently. However, there are areas where further clarification and enhancement could improve readability and comprehension for potential users:
Lines 93-100: This subsection clearly outlines the flexibility of mbctools in handling various data types and scenarios, such as single-end reads and paired-end reads. It may be beneficial to briefly mention examples of sequencing platforms (e.g., Illumina, Ion Torrent) that generate compatible data types, offering a direct reference point for researchers familiar with specific technologies.
Lines 101-107: The requirement for Python3.7 (or higher) and VSEARCH is straightforward, but it could be helpful to link directly to the VSEARCH GitHub page or documentation for users unfamiliar with this tool. Additionally, elaborating on the necessity of Powershell script execution for Windows users could aid in troubleshooting potential installation issues.
Lines 108-132: The detailed setup for directory structure and necessary files is commendable for its thoroughness. However, this section could benefit from:
Simplification: Breaking down this information into bullet points or a checklist might make the setup process seem less daunting.
Visual Aids: Consider including a diagram or flowchart in addition to Figure 3 to visually represent the directory structure and file relationships.
Line 131: The instruction for launching mbctools is clear, but including information about common commands or operations that a new user might need to perform upon starting the tool could enhance usability. A brief "Getting Started" guide within this section, or as a reference to another part of the documentation, would be beneficial.
Software Features
Lines 148-157: This passage effectively outlines the initial data analysis process. Clarifying whether the user has the flexibility to bypass the complete cycle for specific tasks or if they must follow a linear process would be helpful. This could address potential user questions about workflow customizability.
Lines 160-171: The detailed explanation of menu options and the flexibility to refine analysis parameters is commendable. It might be useful to provide examples or case studies where adjusting these parameters significantly impacted the analysis outcome, offering practical insights into how users might leverage these features.(if exists)
Main pipeline description
This section can be used as a supplementary file, rather than being included in the main text, or it can be uploaded to a github page as a document or even a video, which is more valuable to the user.
Conclusion
Lines 398-402: The summary of mbctools’ functionalities is clear and concise. It might be beneficial to briefly recap the main advantages or unique features of mbctools compared to other available tools, reinforcing the reasons for its development and the gaps it aims to fill within the field.
Lines 403-405: Mentioning the customization possibility for the taxonomic assignment step is crucial. It would be helpful to suggest some widely used tools or methods for this purpose, offering a starting point for users less familiar with the options available for taxonomic assignment.
Lines 406-409: The transition towards mentioning the integration with metaXplor is smooth, but expanding on how mbctools specifically enhances data management, visualization, and accessibility through this compatibility could be enriching. Highlighting examples of how this feature has been utilized in previous studies or projects could provide concrete benefits.
Future Directions and Development: While the note on ongoing development hints at mbctools' adaptability, a brief mention of specific areas or features under development could excite potential users about future updates. This could include mentioning planned improvements, new functionalities, or integration with other platforms and tools.
mbctools provides a wrapper that simplifies access to metabarcoding data analyses by providing an easy to install and well describe analysis steps along some options. It relies and depends only on vsearch software which users must download as an executable binary or install from source. It is also notable that mbctools is cross-plateform and can be used as command-line tool and even in HPC environments.
Despite, i suggest authors to improve some points.
1 Users must be informed that they won’t get mbctools working unless they have accessible vsearch installation (via a PATH). This maybe stated in the software Readme section and pypi internet page.
2 Windows users are often less used to command line and software installation procedures. I suggest a well detailed procedure specific to Windows OS.
3 Lines 99/100 : “The pipeline can accommodate a potentially unlimited number of samples”. This is an overstatement. Even more the ability of the mbctools (depending on vsearch) to handle samples depends on the algorithmic complexity and the availability of computational resources. There is no estimation nor data on needed computational resources for processing, for example, the toy dataset. So, authors should this statement or remove it.
4 Line 227, there is a typo on « merge reads »
5 Line 297 for what the “[REF]” stands for? IF it is a reference, please provide it.
Overall, I suggest authors to lighten software features section and add more results from real world or simulated datasets to highlight their tool’s ability on diverse contexts. The detailed version of the “software features” and usage can be then added to the git or any other public repository for future users.
