
Latest recommendations

| Id | Title | Authors | Abstract▲ | Picture | Thematic fields | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
02 Apr 2021

Semi-artificial datasets as a resource for validation of bioinformatics pipelines for plant virus detectionToward a critical assessment of virus detection in plantsRecommended by Hadi Quesneville based on reviews by Alexander Suh and 1 anonymous reviewerThe advent of High Throughput Sequencing (HTS) since the last decade has revealed previously unsuspected diversity of viruses as well as their (sometimes) unexpected presence in some healthy individuals. These results demonstrate that genomics offers a powerful tool for studying viruses at the individual level, allowing an in-depth inventory of those that are infecting an organism. Such approaches make it possible to study viromes with an unprecedented level of detail, both qualitative and quantitative, which opens new venues for analyses of viruses of humans, animals and plants. Consequently, the diagnostic field is using more and more HTS, fueling the need for efficient and reliable bioinformatics tools. Many such tools have already been developed, but in plant disease diagnostics, validation of the bioinformatics pipelines used for the detection of viruses in HTS datasets is still in its infancy. There is an urgent need for benchmarking the different tools and algorithms using well-designed reference datasets generated for this purpose. This is a crucial step to move forward and to improve existing solutions toward well-standardized bioinformatics protocols. This context has led to the creation of the Plant Health Bioinformatics Network (PHBN), a Euphresco network project aiming to build a bioinformatics community working on plant health. One of their objectives is to provide researchers with open-access reference datasets allowing to compare and validate virus detection pipelines. In this framework, Tamisier et al. [1] present real, semi-artificial, and completely artificial datasets, each aimed at addressing challenges that could affect virus detection. These datasets comprise real RNA-seq reads from virus-infected plants as well as simulated virus reads. Such a work, providing open-access datasets for benchmarking bioinformatics tools, should be encouraged as they are key to software improvement as demonstrated by the well-known success story of the protein structure prediction community: their pioneer community-wide effort, called Critical Assessment of protein Structure Prediction (CASP)[2], has been providing research groups since 1994 with an invaluable way to objectively test their structure prediction methods, thereby delivering an independent assessment of state-of-art protein-structure modelling tools. Following this success, many other bioinformatic community developed similar “competitions”, such as RNA-puzzles [3] to predict RNA structures, Critical Assessment of Function Annotation [4] to predict gene functions, Critical Assessment of Prediction of Interactions [5] to predict protein-protein interactions, Assemblathon [6] for genome assembly, etc. These are just a few examples from a long list of successful initiatives. Such efforts enable rigorous assessments of tools, stimulate the developers’ creativity, but also provide user communities with a state-of-art evaluation of available tools. Inspired by these success stories, the authors propose a “VIROMOCK challenge” [7], asking researchers in the field to test their tools and to provide feedback on each dataset through a repository. This initiative, if well followed, will undoubtedly improve the field of virus detection in plants, but also probably in many other organisms. This will be a major contribution to the field of viruses, leading to better diagnostics and, consequently, a better understanding of viral diseases, thus participating in promoting human, animal and plant health. References [1] Tamisier, L., Haegeman, A., Foucart, Y., Fouillien, N., Al Rwahnih, M., Buzkan, N., Candresse, T., Chiumenti, M., De Jonghe, K., Lefebvre, M., Margaria, P., Reynard, J.-S., Stevens, K., Kutnjak, D. and Massart, S. (2021) Semi-artificial datasets as a resource for validation of bioinformatics pipelines for plant virus detection. Zenodo, 4273791, version 4 peer-reviewed and recommended by Peer community in Genomics. doi: https://doi.org/10.5281/zenodo.4273791 [2] Critical Assessment of protein Structure Prediction” (CASP) - https://en.wikipedia.org/wiki/CASP [3] RNA-puzzles - https://www.rnapuzzles.org [4] Critical Assessment of Function Annotation (CAFA) - https://en.wikipedia.org/wiki/Critical_Assessment_of_Function_Annotation [5] Critical Assessment of Prediction of Interactions (CAPI) - https://en.wikipedia.org/wiki/Critical_Assessment_of_Prediction_of_Interactions [6] Assemblathon - https://assemblathon.org [7] VIROMOCK challenge - https://gitlab.com/ilvo/VIROMOCKchallenge | Semi-artificial datasets as a resource for validation of bioinformatics pipelines for plant virus detection | Lucie Tamisier, Annelies Haegeman, Yoika Foucart, Nicolas Fouillien, Maher Al Rwahnih, Nihal Buzkan, Thierry Candresse, Michela Chiumenti, Kris De Jonghe, Marie Lefebvre, Paolo Margaria, Jean Sébastien Reynard, Kristian Stevens, Denis Kutnjak, Séb... | <p>The widespread use of High-Throughput Sequencing (HTS) for detection of plant viruses and sequencing of plant virus genomes has led to the generation of large amounts of data and of bioinformatics challenges to process them. Many bioinformatics... | | Bioinformatics, Plants, Viruses and transposable elements | Hadi Quesneville | 2020-11-27 14:31:47 | ||
18 Feb 2021
Traces of transposable element in genome dark matter co-opted by flowering gene regulation networksUsing small fragments to discover old TE remnants: the Duster approach empowers the TE detectionRecommended by Francois Sabot based on reviews by Josep Casacuberta and 1 anonymous reviewer based on reviews by Josep Casacuberta and 1 anonymous reviewer
Transposable elements are the raw material of the dark matter of the genome, the foundation of the next generation of genes and regulation networks". This sentence could be the essence of the paper of Baud et al. (2021). Transposable elements (TEs) are endogenous mobile genetic elements found in almost all genomes, which were discovered in 1948 by Barbara McClintock (awarded in 1983 the only unshared Medicine Nobel Prize so far). TEs are present everywhere, from a single isolated copy for some elements to more than millions for others, such as Alu. They are founders of major gene lineages (HET-A, TART and telomerases, RAG1/RAG2 proteins from mammals immune system; Diwash et al, 2017), and even of retroviruses (Xiong & Eickbush, 1988). However, most TEs appear as selfish elements that replicate, land in a new genomic region, then start to decay and finally disappear in the midst of the genome, turning into genomic ‘dark matter’ (Vitte et al, 2007). The mutations (single point, deletion, recombination, and so on) that occur during this slow death erase some of their most notable features and signature sequences, rendering them completely unrecognizable after a few million years. Numerous TE detection tools have tried to optimize their detection (Goerner-Potvin & Bourque, 2018), but further improvement is definitely challenging. This is what Baud et al. (2021) accomplished in their paper. They used a simple, elegant and efficient k-mer based approach to find small signatures that, when accumulated, allow identifying very old TEs. Using this method, called Duster, they improved the amount of annotated TEs in the model plant Arabidopsis thaliana by 20%, pushing the part of this genome occupied by TEs up from 40 to almost 50%. They further observed that these very old Duster-specific TEs (i.e., TEs that are only detected by Duster) are, among other properties, close to genes (much more than recent TEs), not targeted by small RNA pathways, and highly associated with conserved regions across the rosid family. In addition, they are highly associated with flowering or stress response genes, and may be involved through exaptation in the evolution of responses to environmental changes. TEs are not just selfish elements: more and more studies have shown their key role in the evolution of their hosts, and tools such as Duster will help us better understand their impact. References Baud, A., Wan, M., Nouaud, D., Francillonne, N., Anxolabéhère, D. and Quesneville, H. (2021). Traces of transposable elements in genome dark matter co-opted by flowering gene regulation networks. bioRxiv, 547877, ver. 5 peer-reviewed and recommended by PCI Genomics.doi: https://doi.org/10.1101/547877 | Traces of transposable element in genome dark matter co-opted by flowering gene regulation networks | Agnes Baud, Mariene Wan, Danielle Nouaud, Nicolas Francillonne, Dominique Anxolabehere, Hadi Quesneville | <p>Transposable elements (TEs) are mobile, repetitive DNA sequences that make the largest contribution to genome bulk. They thus contribute to the so-called 'dark matter of the genome', the part of the genome in which nothing is immediately recogn... | | Bioinformatics, Evolutionary genomics, Functional genomics, Plants, Structural genomics, Viruses and transposable elements | Francois Sabot | Anonymous, Josep Casacuberta | 2020-04-07 17:12:12 | |
06 Jul 2021
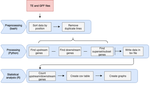
A pipeline to detect the relationship between transposable elements and adjacent genes in host genomesA new tool to cross and analyze TE and gene annotationsRecommended by Emmanuelle Lerat based on reviews by 2 anonymous reviewersTransposable elements (TEs) are important components of genomes. Indeed, they are now recognized as having a major role in gene and genome evolution (Biémont 2010). In particular, several examples have shown that the presence of TEs near genes may influence their functioning, either by recruiting particular epigenetic modifications (Guio et al. 2018) or by directly providing new regulatory sequences allowing new expression patterns (Chung et al. 2007; Sundaram et al. 2014). Therefore, the study of the interaction between TEs and their host genome requires tools to easily cross-annotate both types of entities. In particular, one needs to be able to identify all TEs located in the close vicinity of genes or inside them. Such task may not always be obvious for many biologists, as it requires informatics knowledge to develop their own script codes. In their work, Meguerdichian et al. (2021) propose a command-line pipeline that takes as input the annotations of both genes and TEs for a given genome, then detects and reports the positional relationships between each TE insertion and their closest genes. The results are processed into an R script to provide tables displaying some statistics and graphs to visualize these relationships. This tool has the potential to be very useful for performing preliminary analyses before studying the impact of TEs on gene functioning, especially for biologists. Indeed, it makes it possible to identify genes close to TE insertions. These identified genes could then be specifically considered in order to study in more detail the link between the presence of TEs and their functioning. For example, the identification of TEs close to genes may allow to determine their potential role on gene expression. References Biémont C (2010). A brief history of the status of transposable elements: from junk DNA to major players in evolution. Genetics, 186, 1085–1093. https://doi.org/10.1534/genetics.110.124180 Chung H, Bogwitz MR, McCart C, Andrianopoulos A, ffrench-Constant RH, Batterham P, Daborn PJ (2007). Cis-regulatory elements in the Accord retrotransposon result in tissue-specific expression of the Drosophila melanogaster insecticide resistance gene Cyp6g1. Genetics, 175, 1071–1077. https://doi.org/10.1534/genetics.106.066597 Guio L, Vieira C, González J (2018). Stress affects the epigenetic marks added by natural transposable element insertions in Drosophila melanogaster. Scientific Reports, 8, 12197. https://doi.org/10.1038/s41598-018-30491-w Meguerditchian C, Ergun A, Decroocq V, Lefebvre M, Bui Q-T (2021). A pipeline to detect the relationship between transposable elements and adjacent genes in host genomes. bioRxiv, 2021.02.25.432867, ver. 4 peer-reviewed and recommended by Peer Community In Genomics. https://doi.org/10.1101/2021.02.25.432867 Sundaram V, Cheng Y, Ma Z, Li D, Xing X, Edge P, Snyder MP, Wang T (2014). Widespread contribution of transposable elements to the innovation of gene regulatory networks. Genome Research, 24, 1963–1976. https://doi.org/10.1101/gr.168872.113 | A pipeline to detect the relationship between transposable elements and adjacent genes in host genomes | Caroline Meguerditchian, Ayse Ergun, Veronique Decroocq, Marie Lefebvre, Quynh-Trang Bui | <p>Understanding the relationship between transposable elements (TEs) and their closest positional genes in the host genome is a key point to explore their potential role in genome evolution. Transposable elements can regulate and affect gene expr... | | Bioinformatics, Viruses and transposable elements | Emmanuelle Lerat | 2021-03-03 15:08:34 |




