
Professionalising conservation programmes for local chicken breeds
Trends in genome diversity of small populations under a conservation program: a case study of two French chicken breeds
Abstract
Recommendation: posted 15 September 2024, validated 19 September 2024
Kasper, C. (2024) Professionalising conservation programmes for local chicken breeds. Peer Community in Genomics, 100369. 10.24072/pci.genomics.100369
Recommendation
While it is widely agreed that the conservation of local breeds is key to the maintenance of livestock biodiversity, the implementation of such programmes is often carried out by amateur breeders and may be inadequate due to a lack of knowledge and financial resources. Bortoluzzi et al. (2024) clearly demonstrate the utility of whole-genome sequencing (WGS) data for this purpose, compare two scenarios that differ in the consistency of conservation efforts, and provide valuable recommendations for conservation programmes.
Genetic diversity in livestock is generally considered to be crucial to maintaining food security and ensuring the provision of necessary nutrients to humans (Godde et al. 2021). It is also important to recognise that the preservation of local breeds is a matter of cultural identity for certain regions, and that the products of these breeds are niche products which are in high demand. Especially today, as we face extreme weather conditions, drought and other consequences of global warming, modern breeds selected to perform under constant and temperate conditions are being challenged. The possibility of tapping into the reservoir of genetic variation held by traditional, locally adapted breeds offers an important option for breeding robust livestock. The best way to characterise genetic diversity is through modern molecular methods, based on whole genome sequencing and subsequent advanced population analyses, which has been demonstrated for domesticated and wild chicken (Qanbari et al. 2019).
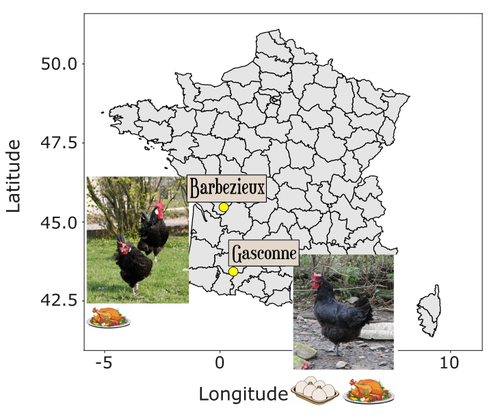
But are local breed conservation programmes up to the task? In their article, Bortoluzzi and colleagues show that well-designed and professionally managed conservation programmes for local chicken breeds are effective in maintaining genetic diversity. Their article is based on a comparison of two examples of conservation programmes for local chicken breeds: the Barbezieux and the Gasconne, which originated from comparably sized founder populations and for which WGS data were available in a biobank at two timepoints, 2003 and 2013, representing 10 generations. While the conservation programme for the former was continuous, that for the latter was interrupted and later started from scratch with a small number of sires and dams.
The greater loss of genomic diversity in the Gasconne than in the Barbezieux shown in this article may therefore be unsurprising, but the authors provide a range of evidence for this using their population genomics toolbox. The less well-managed breed, Gasconne, shows a lower genome-wide heterozygosity, higher lengths of runs of homozygosity, higher levels of genomic inbreeding, a smaller effective population size and a higher genetic load in terms of predicted deleterious mutations.
The sample sizes available for population genetic analyses are typically small for local breeds, which is difficult to change as the populations are very small at any given time. It is therefore all the more important to make the most out of it, and Bortoluzzi and co-authors approach the issue from several angles that help support their claim, using WGS data and the latest genomic resources.
In addition to their analyses, the authors provide clear and valuable advice for the management of such conservation programmes. Their analysis of signatures of selection suggests that, apart from adult fertility, not much selection has been taking place. However, the authors emphasise that clear selection objectives other than maintaining the breed, such as production and product quality, can help conservation efforts by providing better guidelines for managing the programme and increasing the availability of resources for conservation programmes when the products of these local breeds become successful.
In summary, Bortoluzzi et al. (2024) have provided a clear, well-written account of the impact of conservation programme management on the genetic diversity of local chicken breeds, using the most up-to-date genomic resources and analysis methods. As such, this article makes a significant and valuable contribution to the maintenance of genomic resources in livestock, providing approaches and lessons that will hopefully be adopted by other such initiatives.
References
Bortoluzzi C, Restoux G, Rouger R, Desnoues B, Petitjean F, Bosse M, Tixier-Boichard M (2024) Trends in genome diversity of small populations under a conservation program: a case study of two French chicken breeds. bioRxiv, ver. 2 peer-reviewed and recommended by PCI Genomics. https://doi.org/10.1101/2024.02.22.581528
Godde CM, Mason-D’Croz D, Mayberry DE, Thornton PK, Herrero M (2021) Impacts of climate change on the livestock food supply chain; a review of the evidence. Global Food Security 28:100488. https://doi.org/10.1016/j.gfs.2020.100488
Qanbari S, Rubin C-J, Maqbool K, Weigend S, Weigend A, Geibel J, Kerje S, Wurmser C, Peterson AT, IL Brisbin Jr., Preisinger R, Fries R, Simianer H, Andersson L (2019) Genetics of adaptation in modern chicken. PLOS Genetics, 15, e1007989. https://doi.org/10.1371/journal.pgen.1007989

The recommender in charge of the evaluation of the article and the reviewers declared that they have no conflict of interest (as defined in the code of conduct of PCI) with the authors or with the content of the article. The authors declared that they comply with the PCI rule of having no financial conflicts of interest in relation to the content of the article.
European Union’s Horizon 2020 Research and Innovation Programme under Grant Agreement No. 677353.
Evaluation round #1
DOI or URL of the preprint: https://doi.org/10.1101/2024.02.22.581528
Version of the preprint: 1
Author's Reply, 20 Aug 2024
Dear editor,
We have already resubmitted a revised manuscript to biorXiv (Posted August 20, 2024, doi: https://doi.org/10.1101/2024.02.22.581528). We have attached below our responses to the reviewers' comments and remarks.
Best wishes,
Chiara Bortoluzzi, on behalf of all the authors
Decision by Claudia Kasper , posted 26 Apr 2024, validated 26 Apr 2024
, posted 26 Apr 2024, validated 26 Apr 2024
Dear authors,
The manuscript Trends in genome diversity of small populations under a conservation program: a case study of two French chicken breeds has been examined by two expert scientists in population genetics. Although the two reviewers found merit in this study and recognized the quality of the associated paper, they raised a number of concerns that should be addressed before any decision could be rendered. I enclosed below detailed evaluation points. If you think you are able to provide a detailed answer to the different points, I encourage you to respond point by point and submit a new version of the preprint.
Thanks you for submission to PCI Genomics.
Best regards,
Claudia Kasper
Reviewed by Markus Neuditschko, 26 Apr 2024
Bortoluzzi et al., assessed various genetic diversity parameters of two local French Chicken breeds taking advantage of whole-genome sequencing information. The study is well-written and easy to follow. However, I have some major concern about the small sample size, as they only analyzed 15 samples for each time period (2003 and 2013). To better assess the selected sample size, authors should also provide more information about the breeds (e.g. the number of registered animals in the pedigree). Furthermore, the authors did not provide any information, how the 15 animals were selected. Based on pedigree information, it is possible to select most informative individuals by assessing their marginal gene contribution. Besides, that I have also identified some minor issues:
L27: replace still in existence with current livestock populations
L43: Livestock breeders instead of keepers
L45: routinely implemented
L97: robust and generally
Figure 1b: The PCA visualization is not quite informative, as the points are colored according to the time point and not to breed origin. To increase the visualization of the PCA, I suggest to use different shapes (time point) and colors (breeds). Also the variance explained by the first two components is rather low, hence it might also be informative to explore additional components.
L459: The authors mentioned, that the breeds were previously genotyped on 57K SNP chip data and simultaneously highlight the added value of genome sequencing data. To do so, I would suggest to downsize sequencing data to 57K Data, to confirm the arguments posted in this section.
Reviewed by Claudia Fontsere Alemany, 16 Apr 2024
Comments to authors
Bortoluzzi et al present a study on the effects of two different conservation programs on the genomes of two French chicken breeds.
Overall, I find the paper easy to follow and the analysis done appropriate for the research questions they had.
Title and abstract represent the content and the main findings of the study. Introduction is well written and clearly presents the background of the study.
Regarding the results I have a few suggestions that, in my opinion, can enhance the quality of the paper (which I already enjoyed reading):
In all the boxplot comparisons between 2003 and 2013/15, there should be a statistical test to account for significance. Even if the authors describe a trend would be good to add if it is statistically significant or not. I understand that the sample size might be a limitation, but this is something the authors can discuss in the paper.
Also, when using boxplots, it is good practice to also include the individual dots, to get a perception of how they are distributed.
Why did the authors decide to include a semen sample given it is a different sample type? Have the authors detected any difference in number of genotypes detected in this sample compared to the others? Just to check that sample type is not adding more noise.
Why is sample 7218 so different from the rest of 2013/15 Gasconne samples in the PCA? Do you see a different pattern in the other analysis as well? How many of the genotypes are private to this sample? I think this deserves an explanation both in the text (results/discussion) and in the Figure 1 caption.
Regarding the ROH detection, how does your method account for having a window with heterozygous calls inside a ROH that would break it down? Do you account for genotyping errors breaking the (otherwise) long ROH? How do you deal with this? I would like to see a bit more explanations of the method.
Also, in a more theoretical point of view: Does it make sense to calculate ROHs that are <= 100kb? In my previous experience I found them not to be much informative. Is there any reason why the authors decided to include them? On the other hand, I would consider adding another classification of > 10Mb to account for very long ROHs (if in fact they do exist).
Regarding genome-wide heterozygosity, given that the authors already have the regions that are ROHs, I was wondering if they could also plot the heterozygosity outside ROH vs global heterozygosity, to compare both breeds with their historical genome-wide diversity. Of course, I am adding this suggestion only if it adds value to the analysis and discussion.
When discussing over the phenotypic data: could the authors hypothesize why even though there is a loss of heterozygosity and increase of inbreeding, it seems that the Barbezieux chickens are more productive? Would it be possible to link this to a reduction of genetic load? I would like to see a bit more discussion on this, even if it is just ideas/hypothesis.
I want to comment as well that I appreciate the authors adding the positive selection scan section even if they do not find any signature.
I also have just a few more minor comments:
Introduction
Line 27 the authors could add that these 7745 still existing breeds are world-wide (making this fact explicit in the text). Also, is it known how many breeds have gone extinct already in the recent years? If this information is known, could make it even a stronger point for the significance of paper.
There is formatting error in reference (J Fernández, Meuwissen, et al, 2011), in lane 48 (and other places where the reference is added, or the first author is J Fernández) where there is the J of the name in front of the surname.
Material and methods
Line 116. Consider changing Sibs to Siblings wherever this appears.
Line 128. Add (if true) that this is a double-stranded library preparation. Also add citation of the method used.
Lines 136-143. I am curious why the authors decide to do this very conservative strategy (which is ok) of doing SNP calling with different callers and using the overlap. Did you see any weird behavior of any of them. Which one is better?
Lines 145 to 150. Here the authors mention SNPrelate to do the PCA, could the authors add the parameters used or a link to GitHub with the script? Also, for pruning there should be a mention of which software and parameters were used.
Lines157-162. For calculating heterozygosity, the authors cite the paper they have followed. However, in my opinion there should be a bit of explanation of the principles of the methods and/or which software they have used. Also consider adding the script to a GitHub page.
For coverage filtering the authors use 2 times the mean genome-wide coverage. However, in the “Sequencing, read processing and alignment” (see line 143) the authors mention a filtering of 2.5 times the individual mean genome-wide coverage. Probably there is a typo in one of them but just checking.
Line 167: “corrected number of genotypes”. Corrected how?
Line 195. For phasing, the authors use a Ne of 100,000 but later they calculate Ne for its breed (lines 312-313) and the resulting Ne is much smaller. How will this impact your phasing accuracy? Have you considered using the calculated Ne?
Line 201. Pedigree inbreeding: which software did you use to calculate FPED?
Line 205-106. Genome-based inbreeding. Does you “actual length of the genome (Lauto) covered in our dataset” exclude complex regions (duplicates etc)? If so, specify this.
Line 212. Add the link to the whole-genome alignment from Ensembl so it is findable.
Line 227. Add the link to the GERP score so it is findable.
Lines 214-222. Please elaborate a bit more on the parameters and method used for VEP and chCADD – maybe link to the script or just add the parameters used. Also, when describing the filtering criteria: which software and how did you select this?
In formula (1) is written chCADDi = etc. Is the i in the chCADDi=... correct? I am not really used to mathematical notation, but this felt wrong.
Results
Line 275-276. Here the authors put the % of decrease in genome-wide heterozygosity and then in parenthesis the pi value. Is this the pi or the DeltaPi value? Otherwise to which population does this pi value refer to (the pre or the post)? Please clarify this.
Line 286. Add the range of the genome covered with long ROHs for the Gasconne as you have done with the Barbezieux before. “1 to 20 long ROHs that covered up to 29% of the genome (X-X%)”.
Phenotypic data paragraph (lines 343-359). I would love to see supplementary table 5 to 8 as plots (only for supplementary) as well. It is more useful to understand the trends and the data itself.
Discussion
Line 377. The authors mention that the founding nucleus was sampled from fancy breeders, was there an attempt to avoid relatedness? In other words, is it known if the founders were related? In parallel, the authors could estimate relatedness from the 2003 population to see how related they were to start with.
Line 443. First time chCADD acronym appears, define it as chicken CADD as you do here. This happens the first time in line 216.
Lines 458-459. The authors could compare the results of their paper with the previous 57k SNP genotyping. How similar/different are they? Stress better why we need genome-wide sequencing.
Figure and tables
Table 1. I am not familiar with chicken morphology and probably others are not as well. So, it was unclear the value of the “Morphology” column. Is it to show they are “similar breeds”? If that is the case maybe more context could be added in the methods section.
Figure 2. Add “genome-wide” heterozygosity in the first sentence of the caption.
I would recommend the authors to do a statistical test in the heterozygosity levels.
a) What does the values on top of the boxplot represent? DeltaPi? The symbols are strange, and the values are not the same as the ones in the text.
Figure 3.
a) It is difficult to see which individuals are from 2003 and 2013/15 given that bold and italics are complicated to differentiate. The same goes to Gasconne and Barbezieux as some IDs do not have the prefix. I propose to use a color legend for the Heterozygosity bar plot so then it is easier to see which ones are which and add a prefix to each name. Alternatively, you could use color for breed and empty/full bar plot for pre/post sampling.
b) Is the difference statistically significant? Also, add color legend.
c) Add the p-value and Pearson’s r in the caption as well (not just in the text).
Figure 4. Add statistical test for the genetic load.
Supplementary Material
Figure S1. Make the figure bigger (one on top of the other).
I really enjoyed reading and reviewing the paper and looking forward to reading a revised version of it.




