
A protein database to study the origin of metazoans
 based on reviews by Giacomo Mutti and 2 anonymous reviewers
based on reviews by Giacomo Mutti and 2 anonymous reviewers
LukProt: A database of eukaryotic predicted proteins designed for investigations of animal origins
Abstract
Recommendation: posted 26 July 2024, validated 05 August 2024
del Campo, J. (2024) A protein database to study the origin of metazoans. Peer Community in Genomics, 100368. 10.24072/pci.genomics.100368
Recommendation
Sobala (2024) introduces a new, comprehensive, and curated eukaryotic database. It consolidates information from EukProt (Richter et al. 2022) and various other resources to enhance Metazoa representation in existing protein databases. The preprint is of significant interest to the phylogenomics and comparative genomics communities, and I commend the author for their work.
LukProt, the expanded database, significantly increases the taxon sampling within holozoans. It integrates data from the previously assembled EukProt and AniProtDB (Barreira et al. 2021) databases, with additional datasets from early-diverging animal lineages such as ctenophores, sponges, and cnidarians. This effort will undoubtedly be useful for researchers investigating these clades and their origins, as well as for the broader field of comparative genomics.
The author provides both web-portal and command-line versions of the database, making it accessible to users with varying degrees of bioinformatic proficiency. The curation effort is commendable, and I believe the comparative genomics community, especially those interested in animal origins, will find LukProt to be a valuable resource.
References
Barreira SN, Nguyen A-D, Fredriksen MT, Wolfsberg TG, Moreland RT, Baxevanis AD (2021) AniProtDB: A collection of consistently generated metazoan proteomes for comparative genomics studies. Molecular Biology and Evolution 38, 4628–4633. https://doi.org/10.1093/molbev/msab165
Richter DJ, Berney C, Strassert JFH, Poh Y-P, Herman EK, Muñoz-Gómez SA, Wideman JG, Burki F, de Vargas C (2022) EukProt: A database of genome-scale predicted proteins across the diversity of eukaryotes. Peer Community Journal 2, e56. https://doi.org/10.24072/pcjournal.173
Sobala ŁF (2024) LukProt: A database of eukaryotic predicted proteins designed for investigations of animal origins. bioRxiv, ver. 2 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2024.01.30.577650

The recommender in charge of the evaluation of the article and the reviewers declared that they have no conflict of interest (as defined in the code of conduct of PCI) with the authors or with the content of the article. The authors declared that they comply with the PCI rule of having no financial conflicts of interest in relation to the content of the article.
National Science Centre of Poland (NCN) grant no. 2020/36/C/NZ8/00081
Reviewed by anonymous reviewer 2, 26 Jun 2024
Reviewed by Giacomo Mutti , 10 Jun 2024
Reviewed by anonymous reviewer 1, 24 Jun 2024
I would like to thank the author for the work and the update. Now, the phylogenetic methods of the example are much clearer. The analysis of contamination of the proteomes is shared and worth sharing as the user could choose genomes according to contamination thresholds. Moreover, the HGT reasoning, together with the contamination assessment, is more precise now. The documentation shared in the Zenodo repository is better, allowing the user to trace every process step, database file and protein header, which confers robustness to the resource. The new example with the 20 markers is much more suitable for showing the potential of the database and strengthens its power to gain insights into the clades represented in LukProt.
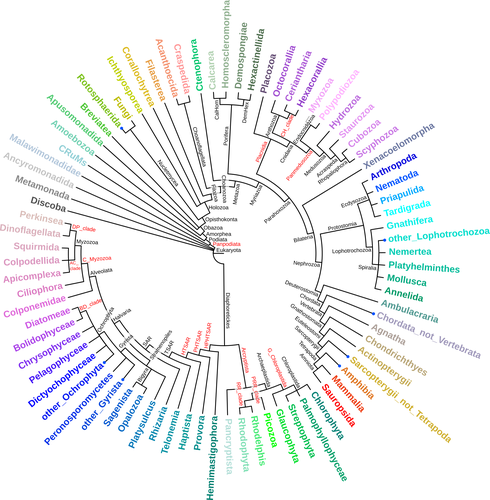
Regarding the colours, it was just a minor comment. They are nice, although I could not distinguish some in my printed version. However, the annotation in the internal nodes helps to detect clades properly. Maybe, and just as a suggestion, reducing the palette to a certain taxonomic level (all SAR with the same colour, not a palette of a given colour) would be a strategy to reduce the colour complexity of the figure and increase the readability, but it is a matter of style.
I consider the reviewed version of the paper suitable for publishing, and the work done has substantially improved the manuscript.
Evaluation round #1
DOI or URL of the preprint: https://doi.org/10.1101/2024.01.30.577650
Version of the preprint: 1
Author's Reply, 28 May 2024
The responses can be found in the PDF file.
The document with tracked changes has the same content as the bioRxiv preprint but the References are in a different font (they needed to be copied and pasted to convert from Zotero to PCI numbered lines, this only works if TrackChange is disabled).
Decision by Javier del Campo, posted 16 Apr 2024, validated 16 Apr 2024
Dear Dr. Sobala,
Despite all the reviewers considering the database that you have developed to be valuable and potentially useful, two of them have some major comments. So, my recommendation would be to revise your preprint following the reviewer's suggestions.
Best regards,
Javier del Campo, PhD
Group Leader, Microbial Ecology and Evolution Laboratory
Chair of the Biodiversity Department
Institut de Biologia Evolutiva (CSIC-UPF)
Passeig Marítim de la Barceloneta 37-49
08003 Barcelona
Reviewed by anonymous reviewer 2, 16 Mar 2024
Reviewed by Giacomo Mutti , 11 Mar 2024
Reviewed by anonymous reviewer 1, 07 Mar 2024
The manuscript of Sobala provides a new pervasive and curated eukaryotic database. It gathers information from EukProt and many other resources to increase the Metazoa sampling in released protein databases. I consider the paper interesting for the phylogenomics and comparative genomics communities, and I thank the author for their work. The analysis and data management are suitable. However, I have found some weaknesses that would make the database user distort the interpretation of the given protein sets. I would recommend this paper for publication after some corrections.
Overview and general comments
The database construction is rigorous as it considers different sources of information, homogenises the IDs, and distributes it using standard formats. The metadata incorporated to the database is easy to read and parse, as well as it is completely integrated with EukProt, maximising the compatibility between both databases. The server that the author provides is accessible, and the taxonomic structure implemented in the server helps the user to perform clade-specific analyses.
I miss an analysis of the contamination and quality of each genome. Providing this information to the users would let them better choose the genomes for downstream analyses. Moreover, this point is essential for lateral gene transfer analyses.
The taxonomy accounts for up-to-date literature, and they performed a readable table, which I personally think is a sound synthesis work and thank. It enforces the deep branches of the events the author considers important for the Metazoan researchers. The author may improve the documentation for the taxonomic tree in the supplementary file. The structure of the taxonomic groups and how they cluster is not clear to me. An indentation table structure with the literature would be more understandable as you would easily identify shallow and deep groups.
Regarding the example analysis, I found the first paragraph in the results more suitable to be written in methods, as the huntingtin example method’s section seems incomplete and unclear. I could not understand why the author included two inference software and which criteria they used for removing branches from the preliminary trees. I emphasise this in the detailed comments of my review.
I finally consider that the assessment of limitations is fair, although I think that the author should add a contamination assessment to the database. BUSCO completeness and contamination values (for instance calculated with OMArk) would be valuable and would make the database even more complete.
Detailed comments on the methods
The methods are proper and suitable for the kind of data that Sobala used. However, I see some weaknesses that they should address:
- Dataset naming: the renaming sounds good. Although the author incorporates the sequence identifier of the original source after the protein ID (L81), a file connecting the protein IDs of LukProt, EukProt, AniProtDB, would be helpful for the community in the cases they change or just for comparison purposes. Despite this, the files are accessible and well-documented in the Zenodo repository.
- Distribution of the database: it may be helpful to separate the BLAST databases by the taxonomic depth of the database in separate compressed files, as Zenodo allows a folder structure. I propose the author to share the folder structure with the compressed version of the database rather than the complete set of databases in a single compressed folder if they consider it appropriate. It would be easier to use and download.
- Data processing: Sobala uses two different software (Trinity and TransAbyss) and multiple versions of the Trinity software for assembling transcriptomes. Moreover, they also use different versions of the software for protein prediction (TransDecoder). However, they do not explain why the author chooses one or the other and the criteria substantiating the software election. The author should include this information in the manuscript. Regarding the clustering, I see the same as previously commented. The author does not specify why and when proteomes are clustered (“in most cases”, L96). Moreover, for the strain “pangenome”, they do not determine whether the CD-HIT parameters are or are not maintained.
- Huntingtin example: as I previously said, the methods section of this example is incomplete. The author should describe better how they obtained the phylogeny with a clear description of each step. They should move the first paragraph of the results section to the methods section with a few changes and clarifications: 1) the procedural scheme is diffuse, I had to read twice to understand the steps they followed; 2) the changes in the CD-HIT clustering identity parameter are not justified; 3) I miss a definition for “outlier” (L192), as some sequences have been removed, I consider necessary to explain which criterion has been used to remove them.
General detailed comments
L58: the author should define AniProtDB, Animal Proteome DataBase (AniProtDB).
L58,64,134,138: AniProtDB appears written differently. The author should homogenise them to the database name in the reference “AniProtDB”.
L127-128: HGT analyses are sensitive to contamination, as they consider that a given protein originated through HGT when it is more similar to a distant taxon than to a close one. For this reason, although I agree with the author that phylogeny will help “drawing conclusions”, I consider that this argument does not apply to HGT. In my opinion, they should at least release a bona fide list of proteins for each organism and a bona fide sister database.
L133: I suppose that a dataset is the whole protein set for a species, but it is not defined. It would be helpful.
L134: a brief comment on the sources of the newly added datasets would be necessary, at least commenting that they have been collected from repositories of numerous studies and the source is available in the metadata table.
L152: the figure 1 colours need to be more different to be easily distinguished. Most of them are similar colours (Apusomonadida, Streptophyta, Breviatea…).
L152: the table 1 title starts with an uppercase letter, while the figure 1 caption starts in lowercase; it should be uppercase.
L202: the figure 2 caption lacks the caption for the panel B, it should be added. Moreover, I suggest to increase the size of the panel B and put the label “A” on top. There are some groups which do not match the topology (Ambulacraria placement, Chordata…), I would understand the size when both topologies match, but I do not see the point here.
L214-216: sequence similarity networks gain insights into deeper relationships and higher detection of far homologs. However, the whole analysis is trying to remove homologs by clustering and manual curation, this sentence may confuse the reader.




