
Latest recommendations

| Id | Title | Authors | Abstract | Picture | Thematic fields▼ | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
24 Feb 2023

MacSyFinder v2: Improved modelling and search engine to identify molecular systems in genomesA unique and customizable approach for functionally annotating prokaryotic genomesRecommended by Gavin Douglas based on reviews by Kwee Boon Brandon Seah and Max Emil Schön based on reviews by Kwee Boon Brandon Seah and Max Emil Schön
Macromolecular System Finder (MacSyFinder) v2 (Néron et al., 2023) is a newly updated approach for performing functional annotation of prokaryotic genomes (Abby et al., 2014). This tool parses an input file of protein sequences from a single genome (either ordered by genome location or unordered) and identifies the presence of specific cellular functions (referred to as “systems”). These systems are called based on two criteria: (1) that the "quorum" of a minimum set of core proteins involved is reached the “quorum” of a minimum set of core proteins being involved that are present, and (2) that the genes encoding these proteins are in the expected genomic organization (e.g., within the same order in an operon), when ordered data is provided. I believe the MacSyFinder approach represents an improvement over more commonly used methods exactly because it can incorporate such information on genomic organization, and also because it is more customizable. Before properly appreciating these points, it is worth noting the norms and key challenges surrounding high-throughput functional annotation of prokaryotic genomes. Genome sequences are being added to online repositories at increasing rates, which has led to an enormous amount of bacterial genome diversity available to investigate (Altermann et al., 2022). A key aspect of understanding this diversity is the functional annotation step, which enables genes to be grouped into more biologically interpretable categories. For instance, gene calls can be mapped against existing Clusters of Orthologous Genes, which are themselves grouped into general categories such as ‘Transcription’ and ‘Lipid metabolism’ (Galperin et al., 2021). This approach is valuable but is primarily used for global summaries of functional annotations within a genome: for example, it could be useful to know that a genome is particularly enriched for genes involved in lipid metabolism. However, knowing that a particular gene is involved in the general process of lipid metabolism is less likely to be actionable. In other words, the desired specificity of a gene’s functional annotation will depend on the exact question being investigated. There is no shortage of functional ontologies in genomics that can be applied for this purpose (Douglas and Langille, 2021), and researchers are often overwhelmed by the choice of which functional ontology to use. In this context, giving researchers the ability to precisely specify the gene families and operon structures they are interested in identifying across genomes provides useful control over what precise functions they are profiling. Of course, most researchers will lack the information and/or expertise to fully take advantage of MacSyFinder’s customizable features, but having this option for specialized purposes is valuable. The other MacSyFinder feature that I find especially noteworthy is that it can incorporate genomic organization (e.g., of genes ordered in operons) when calling systems. This is a rare feature among commonly used tools for functional annotation and likely results in much higher specificity. As the authors note, this capability makes the co-occurrence of paralogs, and other divergent genes that share sequence similarity, to contribute less noise (i.e., they result in fewer false positive calls). It is important to emphasize that these features are not new additions in MacSyFinder v2, but there are many other valuable changes. Most practically, this release is written in Python 3, rather than the obsolete Python 2.7, and was made more computationally efficient, which will enable MacSyFinder to be more widely used and more easily maintained moving forward. In addition, the search algorithm for analyzing individual proteins was fundamentally updated as well. The authors show that their improvements to the search algorithm result in an 8% and 20% increase in the number of identified calls for single and multi-locus secretion systems, respectively. Taken together, MacSyFinder v2 represents both practical and scientific improvements over the previous version, which will be of great value to the field. References Abby SS, Néron B, Ménager H, Touchon M, Rocha EPC (2014) MacSyFinder: A Program to Mine Genomes for Molecular Systems with an Application to CRISPR-Cas Systems. PLOS ONE, 9, e110726. https://doi.org/10.1371/journal.pone.0110726 Altermann E, Tegetmeyer HE, Chanyi RM (2022) The evolution of bacterial genome assemblies - where do we need to go next? Microbiome Research Reports, 1, 15. https://doi.org/10.20517/mrr.2022.02 Douglas GM, Langille MGI (2021) A primer and discussion on DNA-based microbiome data and related bioinformatics analyses. Peer Community Journal, 1. https://doi.org/10.24072/pcjournal.2 Galperin MY, Wolf YI, Makarova KS, Vera Alvarez R, Landsman D, Koonin EV (2021) COG database update: focus on microbial diversity, model organisms, and widespread pathogens. Nucleic Acids Research, 49, D274–D281. https://doi.org/10.1093/nar/gkaa1018 Néron B, Denise R, Coluzzi C, Touchon M, Rocha EPC, Abby SS (2023) MacSyFinder v2: Improved modelling and search engine to identify molecular systems in genomes. bioRxiv, 2022.09.02.506364, ver. 2 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2022.09.02.506364 | MacSyFinder v2: Improved modelling and search engine to identify molecular systems in genomes | Bertrand Néron, Rémi Denise, Charles Coluzzi, Marie Touchon, Eduardo P. C. Rocha, Sophie S. Abby | <p style="text-align: justify;">Complex cellular functions are usually encoded by a set of genes in one or a few organized genetic loci in microbial genomes. Macromolecular System Finder (MacSyFinder) is a program that uses these properties to mod... | | Bacteria and archaea, Bioinformatics, Functional genomics | Gavin Douglas | Kwee Boon Brandon Seah, Max Emil Schön | 2022-09-09 10:30:31 | |
07 Feb 2023
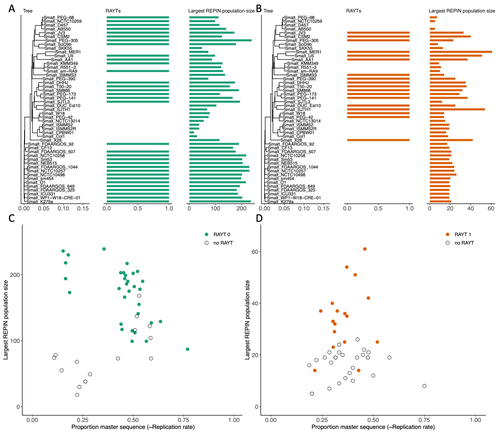
RAREFAN: A webservice to identify REPINs and RAYTs in bacterial genomesA workflow for studying enigmatic non-autonomous transposable elements across bacteriaRecommended by Gavin Douglas based on reviews by Sophie Abby and 1 anonymous reviewer
Repetitive extragenic palindromic sequences (REPs) are common repetitive elements in bacterial genomes (Gilson et al., 1984; Stern et al., 1984). In 2011, Bertels and Rainey identified that REPs are overrepresented in pairs of inverted repeats, which likely form hairpin structures, that they referred to as “REP doublets forming hairpins” (REPINs). Based on bioinformatics analyses, they argued that REPINs are likely selfish elements that evolved from REPs flanking particular transposes (Bertels and Rainey, 2011). These transposases, so-called REP-associated tyrosine transposases (RAYTs), were known to be highly associated with the REP content in a genome and to have characteristic upstream and downstream flanking REPs (Nunvar et al., 2010). The flanking REPs likely enable RAYT transposition, and their horizontal replication is physically linked to this process. In contrast, Bertels and Rainey hypothesized that REPINs are selfish elements that are highly replicated due to the similarity in arrangement to these RAYT-flanking REPs, but independent of RAYT transposition and generally with no impact on bacterial fitness (Bertels and Rainey, 2011). This last point was especially contentious, as REPINs are highly conserved within species (Bertels and Rainey, 2023), which is unusual for non-beneficial bacterial DNA (Mira et al., 2001). Bertels and Rainey have since refined their argument to be that REPINs must provide benefits to host cells, but that there are nonetheless signatures of intragenomic conflict in genomes associated with these elements (Bertels and Rainey, 2023). These signatures reflect the divergent levels of selections driving REPIN distribution: selection at the level of each DNA element and selection on each individual bacterium. I found this observation particularly interesting as I and my colleague recently argued that these divergent levels of selection, and the interaction between them, is key to understanding bacterial pangenome diversity (Douglas and Shapiro, 2021). REPINs could be an excellent system for investigating these levels of selection across bacteria more generally. The problem is that REPINs have not been widely characterized in bacterial genomes, partially because no bioinformatic workflow has been available for this purpose. To address this problem, Fortmann-Grote et al. (2023) developed RAREFAN, which is a web server for identifying RAYTs and associated REPINs in a set of input genomes. The authors showcase their tool by applying it to 49 Stenotrophomonas maltophilia genomes and providing examples of how to identify and assess RAYT-REPIN hits. The workflow requires several manual steps, but nonetheless represents a straightforward and standardized approach. Overall, this workflow should enable RAYTs and REPINs to be identified across diverse bacterial species, which will facilitate further investigation into the mechanisms driving their maintenance and spread. References Bertels F, Rainey PB (2023) Ancient Darwinian replicators nested within eubacterial genomes. BioEssays, 45, 2200085. https://doi.org/10.1002/bies.202200085 Bertels F, Rainey PB (2011) Within-Genome Evolution of REPINs: a New Family of Miniature Mobile DNA in Bacteria. PLOS Genetics, 7, e1002132. https://doi.org/10.1371/journal.pgen.1002132 Douglas GM, Shapiro BJ (2021) Genic Selection Within Prokaryotic Pangenomes. Genome Biology and Evolution, 13, evab234. https://doi.org/10.1093/gbe/evab234 Fortmann-Grote C, Irmer J von, Bertels F (2023) RAREFAN: A webservice to identify REPINs and RAYTs in bacterial genomes. bioRxiv, 2022.05.22.493013, ver. 4 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2022.05.22.493013 Gilson E, Clément J m., Brutlag D, Hofnung M (1984) A family of dispersed repetitive extragenic palindromic DNA sequences in E. coli. The EMBO Journal, 3, 1417–1421. https://doi.org/10.1002/j.1460-2075.1984.tb01986.x Mira A, Ochman H, Moran NA (2001) Deletional bias and the evolution of bacterial genomes. Trends in Genetics, 17, 589–596. https://doi.org/10.1016/S0168-9525(01)02447-7 Nunvar J, Huckova T, Licha I (2010) Identification and characterization of repetitive extragenic palindromes (REP)-associated tyrosine transposases: implications for REP evolution and dynamics in bacterial genomes. BMC Genomics, 11, 44. https://doi.org/10.1186/1471-2164-11-44 Stern MJ, Ames GF-L, Smith NH, Clare Robinson E, Higgins CF (1984) Repetitive extragenic palindromic sequences: A major component of the bacterial genome. Cell, 37, 1015–1026. https://doi.org/10.1016/0092-8674(84)90436-7 | RAREFAN: A webservice to identify REPINs and RAYTs in bacterial genomes | Frederic Bertels, Julia von Irmer, Carsten Fortmann-Grote | <p style="text-align: justify;">Compared to eukaryotes, repetitive sequences are rare in bacterial genomes and usually do not persist for long. Yet, there is at least one class of persistent prokaryotic mobile genetic elements: REPINs. REPINs are ... | | Bacteria and archaea, Bioinformatics, Evolutionary genomics, Viruses and transposable elements | Gavin Douglas | 2022-06-07 08:21:34 | ||
08 Apr 2022

POSTPRINT
Phylogenetics in the Genomic Era“Phylogenetics in the Genomic Era” brings together experts in the field to present a comprehensive synthesisRecommended by Robert Waterhouse and Karen MeusemannE-book: Phylogenetics in the Genomic Era (Scornavacca et al. 2021) This book was not peer-reviewed by PCI Genomics. It has undergone an internal review by the editors. Accurate reconstructions of the relationships amongst species and the genes encoded in their genomes are an essential foundation for almost all evolutionary inferences emerging from downstream analyses. Molecular phylogenetics has developed as a field over many decades to build suites of models and methods to reconstruct reliable trees that explain, support, or refute such inferences. The genomic era has brought new challenges and opportunities to the field, opening up new areas of research and algorithm development to take advantage of the accumulating large-scale data. Such ‘big-data’ phylogenetics has come to be known as phylogenomics, which broadly aims to connect molecular and evolutionary biology research to address questions centred on relationships amongst taxa, mechanisms of molecular evolution, and the biological functions of genes and other genomic elements. This book brings together experts in the field to present a comprehensive synthesis of Phylogenetics in the Genomic Era, covering key conceptual and methodological aspects of how to build accurate phylogenies and how to apply them in molecular and evolutionary research. The paragraphs below briefly summarise the five constituent parts of the book, highlighting the key concepts, methods, and applications that each part addresses. Being organised in an accessible style, while presenting details to provide depth where necessary, and including guides describing real-world examples of major phylogenomic tools, this collection represents an invaluable resource, particularly for students and newcomers to the field of phylogenomics. Part 1: Phylogenetic analyses in the genomic era Modelling how sequences evolve is a fundamental cornerstone of phylogenetic reconstructions. This part of the book introduces the reader to phylogenetic inference methods and algorithmic optimisations in the contexts of Markov, Maximum Likelihood, and Bayesian models of sequence evolution. The main concepts and theoretical considerations are mapped out for probabilistic Markov models, efficient tree building with Maximum Likelihood methods, and the flexibility and robustness of Bayesian approaches. These are supported with practical examples of phylogenomic applications using the popular tools RAxML and PhyloBayes. By considering theoretical, algorithmic, and practical aspects, these chapters provide readers with a holistic overview of the challenges and recent advances in developing scalable phylogenetic analyses in the genomic era. Part 2: Data quality, model adequacy This part focuses on the importance of considering the appropriateness of the evolutionary models used and the accuracy of the underlying molecular and genomic data. Both these aspects can profoundly affect the results when applying current phylogenomic methods to make inferences about complex biological and evolutionary processes. A clear example is presented for methods for building multiple sequence alignments and subsequent filtering approaches that can greatly impact phylogeny inference. The importance of error detection in (meta)barcode sequencing data is also highlighted, with solutions offered by the MACSE_BARCODE pipeline for accurate taxonomic assignments. Orthology datasets are essential markers for phylogenomic inferences, but the overview of concepts and methods presented shows that they too face challenges with respect to model selection and data quality. Finally, an innovative approach using ancestral gene order reconstructions provides new perspectives on how to assess gene tree accuracy for phylogenomic analyses. By emphasising through examples the importance of using appropriate evolutionary models and assessing input data quality, these chapters alert readers to key limitations that the field as a whole strives to address. Part 3: Resolving phylogenomic conflicts Conflicting phylogenetic signals are commonplace and may derive from statistical or systematic bias. This part of the book addresses possible causes of conflict, discordance between gene trees and species trees and how processes that lead to such conflicts can be described by phylogenetic models. Furthermore, it provides an overview of various models and methods with examples in phylogenomics including their pros and cons. Outlined in detail is the multispecies coalescent model (MSC) and its applications in phylogenomics. An interesting aspect is that different phylogenetic signals leading to conflict are in fact a key source of information rather than a problem that can – and should – be used to point to events like introgression or hybridisation, highlighting possible future trends in this research area. Last but not least, this part of the book also addresses inferring species trees by concatenating single multiple sequence alignments (gene alignments) versus inferring the species tree based on ensembles of single gene trees pointing out advantages and disadvantages of both approaches. As an important take home message from these chapters, it is recommended to be flexible and identify the most appropriate approach for each dataset to be analysed since this may tremendously differ depending on the dataset, setting, taxa, and phylogenetic level addressed by the researcher. Part 4: Functional evolutionary genomics In this part of the book the focus shifts to functional considerations of phylogenomics approaches both in terms of molecular evolution and adaptation and with respect to gene expression. The utility of multi-species analysis is clearly presented in the context of annotating functional genomic elements through quantifying evolutionary constraint and protein-coding potential. An historical perspective on characterising rates of change highlights how phylogenomic datasets help to understand the modes of molecular evolution across the genome, over time, and between lineages. These are contextualised with respect to the specific aim of detecting signatures of adaptation from protein-coding DNA alignments using the example of the MutSelDP-ω∗ model. This is extended with the presentation of the generally rare case of adaptive sequence convergence, where consideration of appropriate models and knowledge of gene functions and phenotypic effects are needed. Constrained or relaxed, selection pressures on sequence or copy-number affect genomic elements in different ways, making the very concept of function difficult to pin down despite it being fundamental to relate the genome to the phenotype and organismal fitness. Here gene expression provides a measurable intermediate, for which the Expression Comparison tool from the Bgee suite allows exploration of expression patterns across multiple animal species taking into account anatomical homology. Overall, phylogenomics applications in functional evolutionary genomics build on a rich theoretical history from molecular analyses where integration with knowledge of gene functions is challenging but critical. Part 5: Phylogenomic applications Rather than attempting to review the full extent of applications linked to phylogenomics, this part of the book focuses on providing detailed specific insights into selected examples and methods concerning i) estimating divergence times, and ii) species delimitation in the era of ‘omics’ data. With respect to estimating divergence times, an exemplary overview is provided for fossil data recovered from geological records, either using fossil data as calibration points with an extant-species-inferred phylogeny, or using a fossilised birth-death process as a mechanistic model that accounts for lineage diversification. Included is a tutorial for a joint approach to infer phylogenies and estimate divergence times using the RevBayes software with various models implemented for different applications and datasets incorporating molecular and morphological data. An interesting excursion is outlined focusing on timescale estimates with respect to viral evolution introducing BEAGLE, a high-performance likelihood-calculation platform that can be used on multi-core systems. As a second major subject, species delimitation is addressed since currently the increasing amount of available genomic data enables extensive inferences, for instance about the degree of genetic isolation among species and ancient and recent introgression events. Describing the history of molecular species delimitation up to the current genomic era and presenting widely used computational methods incorporating single- and multi-locus genomic data, pros and cons are addressed. Finally, a proposal for a new method for delimiting species based on empirical criteria is outlined. In the closing chapter of this part of the book, BPP (Bayesian Markov chain Monte Carlo program) for analysing multi-locus sequence data under the multispecies coalescent (MSC) model with and without introgression is introduced, including a tutorial. These examples together provide accessible details on key conceptual and methodological aspects related to the application of phylogenetics in the genomic era. References Scornavacca C, Delsuc F, Galtier N (2021) Phylogenetics in the Genomic Era. https://hal.inria.fr/PGE/ | Phylogenetics in the Genomic Era | Céline Scornavacca, Frédéric Delsuc, Nicolas Galtier | <p style="text-align: justify;">Molecular phylogenetics was born in the middle of the 20th century, when the advent of protein and DNA sequencing offered a novel way to study the evolutionary relationships between living organisms. The first 50 ye... | | Bacteria and archaea, Bioinformatics, Evolutionary genomics, Functional genomics, Fungi, Plants, Population genomics, Vertebrates, Viruses and transposable elements | Robert Waterhouse | 2022-03-15 17:43:52 |




