
Recommendation
 based on reviews by 2 anonymous reviewers
based on reviews by 2 anonymous reviewers
The first time I heard of ”onagres” in French was when I was a teenager, through the books of Pierre Bordage as fantastic monsters, or through historical games as Roman siege weapons (onagers). At this time, I was far from imagining that “onagre” also refers to a very large flowering plant family, as it is the French term for evening primroses.
In this family, the genus Ludwigia comprises species that are invasive (resembling in that way the ancient armies using onagers to invade cities) in aquatic environments, degrading ecosystems already fragilized by human activities. To counteract this phenomenon, it is of high importance to understand their propagation of these species. However, the knowledge about their genetics and diversity is very scarce, and thus tracking their dispersal using genetic information is complicated, and in fact almost impossible.
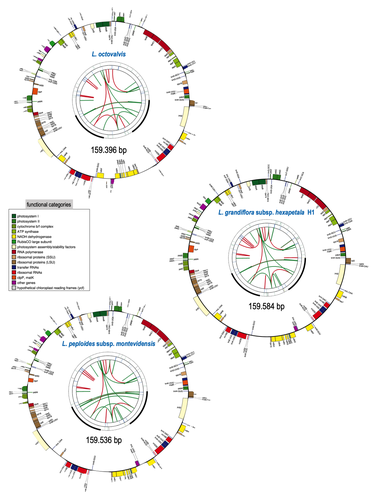
Barloy-Hubler et al. (2024) proposed in the present manuscript a new set of chloroplastic genomes from two of these species, Ludwigia grandiflora subsp. hexapetala and Ludwigia peploides subsp. montevidensis, and compared them to the published chloroplastic genome of Ludwigia octovalis. They explored the possibility of assembling these genomes relying solely on short reads and showed that long reads were necessary to obtain an almost complete assembly for these plastid genomes. In addition, through this approach, they detected two haplotypes in Ludwigia grandiflora subsp. hexapetala as compared to one in a short-read assembly. This highlights the need for long reads data to assess the structure and diversity of chloroplastic genomes. The authors were also able to clarify the phylogeny of the genus Ludwigia. Finally, they identified multiple potential single nucleotide polymorphisms and simple sequence repeats for future evaluation of diversity and dispersal of those invasive species.
This analysis, while appearing more technical than biological at first glance, is in fact of high importance for the understanding of ecology and preservation of fragile ecosystems, such as the European watersheds. Indeed, new scientific results and insights are generally linked to a reevaluation of previously analyzed data or samples through new technologies, and this paper is a quite clever example of that matter.
References
Barloy-Hubler F, Gac A-LL, Boury C, Guichoux E, Barloy D (2024) Sequencing, de novo assembly of Ludwigia plastomes, and comparative analysis within the Onagraceae family. bioRxiv, ver. 5 peer-reviewed and recommended by PCI Genomics. https://doi.org/10.1101/2023.10.20.563230
Bordage, P (1993) Les Guerriers du Silence, L'Atalante, ISBN 9782905158697
DOI or URL of the preprint: https://doi.org/10.1101/2023.10.20.563230
Version of the preprint: 4
Dear Dr Sabot,
Thank you for your positive feedback.
As requested, we have submitted the sequencing data to the EBI-ENA database and included the accession numbers in the text on lines 661-665 (highlighted in blue).
The article has been resubmitted to bioRxiv and we are awaiting your final recommendation.
Best regards,
Dominique Barloy for authors
, posted 27 Nov 2024, validated 28 Nov 2024Dear Colleagues
First of all I apologize for the long delay in validating the manuscript, I was overwhelmed and did not accurately fixed my working time...
However, I have been through the answers to reviewers and they are ok for me, but for one which is mandatory for PCI: "Data available on request" is not a OpenScience-complient statement and the data must be available in a dedicated database (EBI e.g.)
If the data cannot be released at the same time as the paper, without restrictions outside of citations, we cannot accept the paper in PCI.
Once the release be performed, I will accept the paper.
Sincerely yours,
Francois Sabot
DOI or URL of the preprint: https://doi.org/10.1101/2023.10.20.563230
Version of the preprint: 3
Please find in tr&acked changes document only tables and figures changed as request by referees. All table and figure are in main document in bioXriv.
We apologize for this
, posted 05 Sep 2024, validated 05 Sep 2024Dear Colleagues
Thanks first for the time you already spent on the modification of your work in this second round of review. You answered most of the questions of the reviewers and debates were nice.
However, some questions/missing informations are still lacking in the current version (as pointed out by reviewer 1) or some minor modifications requested by reviewer 2.
Would you be able to modify the manuscript version according these requests ? If so I would be able to recommand it quite quickly
Sincerely yours
Francois Sabot
First I would like to thank the authors for answering all my questions. Although I may not necessarily agree with their responses, I respect their point of view, and this does not diminish the quality of their work. Therefore, I will not pursue these discussions further. However, I still have a few questions:
Material and & methods
(1) Did the authors create a museum voucher for the two samples they sequenced? I understand that the samples have been destroyed, but it is common practice to deposit a sample in a museum before sequencing a new reference genome, so it can serve as a reference for future taxonomic (re)identification. If this was done, please provide the voucher number and the name of the museum.
(2) How were the short reads filtered? If the authors used the default parameters of fastp (not explicitly mentioned, but assumed based on the use of default settings for other software), the minimum quality threshold was 15, which seems rather low to me.
Results
Plastome short read assembly.
(3) Please provide the total number of raw reads obtained and the mean quality before filtering. This information will help determine whether the number of chloroplast reads extracted is reasonable.
(4) In the Methods section, the authors mentioned producing two preliminary draft assemblies of Lgh, one with GetOrganelle and one with NOVOPlasty. Here, they state that they used the GetOrganelle draft to extract chloroplast short reads. What about the NOVOPlasty draft?
Plastome long read assembly.
(5) Please provide some metrics (particularly the N50) to assess the quality of the sequencing.
Annotation
(6) The authors performed a new annotation of the Lo plastome. What differences were observed between the two annotations? Was the new annotation submitted to GenBank?
(7) For Figure 3, please include the GenBank accession number directly on each graph.
Discussion
(8) Lines 547-519. Please cite some papers where this has been demonstrated previously. As currently written, the sentence implies that you discovered that hybrid assembly is superior to SR or LR assemblies alone, but others have already published similar findings before. If you prefer not to cite previous work, you should at least consider rephrasing the sentence to: « After conducting our research, we discovered that hybrid assembly, which incorporates both long and short read sequences, resulted in the most superior complete assemblies of Lgh plastome. »
(9) Line 526-528. I disagree with the statement that it was impossible to identify both haplotypes because only LR were available. In fact, LR should be the most suitable sequences for identifying both haplotypes, as they are the only ones capable of covering the SSC and its boundaries. When I used ptGAUL with LR alone, I successfully identified both haplotypes, provided I had sufficient raw reads. Please note that I am not the author of this software and have no relations with its developers.
In your LR assembly results for Lgh, you obtained haplotype 2 with CANU and haplotype 1 with FLYE. The correction with SR did not change the haplotype. It appears that, in your case, the haplotype identified may depend on the software used or on reconstructing one haplotype or the other by chance, rather than on whether SR correction is applied. Therefore, unless you can demonstrate that SR are absolutely necessary to observe both haplotypes, I suggest revising this statement.
Conclusion
(10) Line 643. Be careful, you did not observe two haplotypes for Lpm.
Figures and Tables
(11) Table 1. Please replace “;” by “,” for bp and “;” by “.” for %.
(12) Table 2-3 + all the text. Gene names should be italicized, except for capital letters. Eg. psaB → psaB (psa = italic; B = non italic). Please ensure that this formatting is correct for every occurrence of a gene name throughout the text.
Data availability
(13) I understand that a whole genome analysis is underway, but this should not prevent the publication of chloroplast data alone (for example, the files obtained after filtering chloroplast reads with the draft assembly).
DOI or URL of the preprint: https://doi.org/10.1101/2023.10.20.563230
Version of the preprint: 2
Rennes, France, July 16, 2024
Dear PCI Evolutionary Biology Recommender and reviewers,
Thanks to have considered our paper previously entitled “Sequencing, de novo assembly of Ludwigia plastomes, and comparative analysis within the Onagraceae family”
All remarks and clarifications asked by the reviewers have been taken into account and each modification compared with the first manuscript is highlighted in blue in the revised manuscript. We thank the reviewers for pointing out the need to rephrase and clarify some aspects of the paper.
We are pleased to submit the reviewed manuscript, which was significantly improved after responding to all reviewer comments and providing a point-by-point reply to each remark below.
Yours sincerely,
For the authors,
Dominique Barloy
Review by anonymous reviewer 1, 05 Feb 2024 15:37
In this manuscript, the authors describe the chloroplast genome’s reconstruction of two species from Ludwigia genus. They used several approaches: short-read, long-read and long-read with correction using short-reads. They concluded that the hybrid method is the most efficient. Later, they described the composition of the genomes (number of genes, number and position of SSR, position of IR boundaries, etc.) and produced a phylogeny of the family based on one gene, matK.
The introduction is not very attractive. As the species is not well known, its description and a justification of its interest should be mentionned in the first paragraph.
Answer: Thank you for this remark.
Action: We have added paragraphs (blue underlined) to better describe Ludwigia and its associated challenges in the Abstract (lines 14-20) and Introduction (lines 58-61; 68-70; 75-79).
The material and methods is written in a very confused way and it is frequently difficult to understand what the authors have done exactly. For example, the authors said “Chloroplastic R1 and R2 reads were used with and without prior error correction using ONT reads with BayesHammer”.
Answer: Thank you for this remark.
Action: Chloroplast assemblies section has been rewritten in order to clarify the strategy in Materials and methods (lines 200-212) as well as in Results (lines 272-290).
The whole process of genome reconstruction is unclear. As I understand, the authors reconstructed a first sequence using short-reads. Then they used this 1rst sequence to filter chloroplast reads and reconstruct four sequences using short-reads (using ABySS, MEGAHIT, Velvet and SPAdes ) and two sequences using long-reads (using FLYE and CANU). What’s the point of this two steps reconstruction?
Answer: The objective is to compare the performance of assemblers using only short reads, those using only long reads, and hybrid assemblers, as in most benchmarking experiments. This is a standard process in bioinformatics and is important for this new plant models.
Why not using an existing whole chloroplast sequence to filter chloroplast reads then to reconstruct only two new sequences (one using short-reads, one using long-reads)?
Answer: We initially performed chloroplastic sequence selection using the only chloroplast available at the time of the study (L. octovalvis), but the results were better and more accurate using the draft genomes produced using GetOrganelle on L. grandiflora reads. Therefore, the initial strategy was not retained and not detailed in the manuscript. Regarding the second comment, assembling only one SR sequence and one LR sequence is not feasible without benchmarking and selecting the best tool for our models (which we did in the article), and it would have also deprived us of the hybrid assembly.
I also wonder why presenting and comparing five different methods of reconstructions instead of the only one that was used to generate the sequences that are actually submitted to Genbank. In a technical paper talking about comparison of assembly software, I would understand, here I would rather read something more concise and focused.
Answer: Our study highlights the importance of algorithm selection on assembly outcomes and emphasizes how crucial this choice is for obtaining the most accurate and comprehensive genome possible. From our experience, benchmarking is essential because different tools yield different results depending on genomic complexity (such as repeat number, ploidy, and intraspecific variability). For this reason, we consider benchmarking not merely a matter of technique but more as scientific rigor.
Action: However, in order to take this comment into consideration, we have condensed the paragraphs “ Plastome short read assembly” and “Plastome long read assembly” in the results section (blue underlined)
The description of phylogeny reconstruction is confusing as well. “We reconstructed phylogenetic relationships among plastomes of Onagraceae. The FFT-NS-2 method in MAFFT 7 [55] was used to align all plastomes with one of the IRs removed to avoid data duplication.” suggests that the phylogeny was done using whole plastome sequences, while “we propose a phylogenetic tree from Ludwigia matK sequences (Figure 8).” suggests that the phylogeny was reconstructed using only one gene, more precisely only 149 amino acids.
Answer: Thank you for bringing this to our attention, there is indeed an error with this paragraph.
Action: The corresponding Materials and Methods paragraph has been rewritten and renamed "Phylogenetic analysis of Ludwigia based on matK sequences" (lines 254-263).
The results and discussion are many times oversold.
“After conducting our research, we discovered that hybrid assembly, which incorporates both long and short read sequences, resulted in the most superior complete assemblies. This innovative approach capitalizes on the advantages of both sequencing technologies, harnessing the accuracy of short read sequences and the length of long read sequences.”. It seems that the authors discovered that hybrid assembly is more efficient. I’m surprised as this is well known (see for example Jain et al. Nature Biotech 2018 or Mak et al. MBE 2023) and already incorporated on chloroplast assembly pipeline (ex. ptGAUL, Zhou et al. Mol Ecol Res 2023).
Answer: Our statement was inadequately expressed as out observation about hybrid assembly holds true for our study and correlates with findings from other similar studies. However, we noted that among the 8283 RefSeq plastomes (current release from GenBank) for which we have sequencing information, 98.1% originate from short read sequences sequencing, 0.7% from long reads sequencing, and only 1.2% from hybrid assemblies. For this reason, we find it important to highlight that, in our study, hybrid assembly has demonstrated notable performance. Regarding ptGAUL, this tool is recent and was not available at the time of our study.
Action: The sentence regarding hybrid assembly has been modified to accommodate this comment (lines 521-523).
Same comment about the flip-flop organization of the LSC. The existence of both haplotypes has already been described. This study adds one more species to the list of species where flip-flop organization was described, but it does not justify the paragraph in the discussion.
Answer: Yes, we agree, as stated in our discussion paragraph. Existence of two haplotypes is cited in the literature, and our study is not a "first." However, it is the first time that two haplotypes have been confirmed and described in Ludwigia and in Onagraceae, through the use of long reads and hybrid assembly. Moreover, the vast majority of RefSeq plastomes are still described as a single haplotype, raising questions about the connection between the very low proportion of plastomes derived from long read and/or hybrid assembly (cf previous response) and the scant description of multiple haplotypes. These are the two points we wanted to emphasize in the discussion.
Action: The corresponding discussion paragraph was rewritten in order to clarify these statements (lines 524-541)
Similar comment about the evolution of IR. Despite being interesting, I’m not sure this discussion has its place on a paper announcing a new chloroplast sequence. This part deserves to be more developed and published alone, rather than buried in a technical paper.
Answer: We found only one sentence in the discussion concerning the IR and mentioning the GC content, indicating that our results are in agreement with the literature (lines 552-555). Therefore, we do not understand and can’t address this comment.
Another important point is the absence of code lines used to analyse the data. Raw data (Illumina and ONT) are not provided. Without this information, the analysis is not reproducible. Similarly, the authors did not mention the references of the samples used for the phylogeny.
Answer/Action: The lines of code were not included because all the tools were used with default parameters (unless otherwise specified). However, we realized this was not clearly indicated and have added it to the Materials and Methods section (line 217). This additional information ensures the study is easily reproducible. Regarding the raw data, they are not yet available online as the nuclear genome is still being assembled by a PhD student. However, we understand the reviewer’s comment and have added a sentence with this indication (line 654) indicating that the chloroplastic SR and LR are available upon request. As for the references used in phylogeny, they have been added Figure 8.
Review by anonymous reviewer 2, 07 May 2024 03:16
Abstract
The abstract provides a concise overview of the study and key findings. However, it might be
further improved by clarifying the importance of the study:
- Explain why these Ludwigia species are important.
Action: following sentences were added. (lines 14-20)
- Provide which methods or specific sequencing technologies were used to generate the plastid genomes.
Action: sentences changed (lines 22-23 and 25-26)
- Provide numerical information on the plastid genome size of three Ludwigia species.
Action: done (lines 27-28)
Line 12-13: I suggest split this sentence in two: “The Onagraceae family, which belongs to
the order Myrtales, consists of approximately 657 species and 17 genera, including the
genus Ludwigia L., which is comprised of 82 species.”
Action: Done (lines 46-50)
Introduction
What is the importance of Ludwigia species?
- I suggest including 1-2 sentences in the introduction to highlight the importance of these species.
Answer: Thank you for your suggestion; we have incorporated additional sentences addressing this point.
Action: see lines 58-61
- Additionally, provide a more detailed rationale for the importance of studying plastid genomes in Ludwigia species.
Answer : Thank you for this feedback. We added additional sentences about this point.
Action: see lines 68-70; 75-79
Line 44, 107: Ludwigia needs to be italicized. Please check throughout the manuscript
carefully.
Action: Done
Line 63: Add Reference.
Answer: The reference corresponds to the GenBank release used during the study (Genbank Release 255: April 15 2023)
Action: It has been duly included in the text (line 80).
Line 67: Add Reference. This is the same reference as previously answered (Genbank Release 255: April 15, 2023) and that was added in the text (line 81)
Line 95-97: The sentence seems not complete “Recent long-read sequencing (> 1000 bp)
provides strong evidence that terrestrial plant plastomes have two structural haplotypes,
present in equal proportions and differing in IR orientation of the [22].
Action: Thanks for this observation. Sentence was changed (lines 113-114)
Materials and methods
- Why did authors not perform short-read Illumina sequencing and long-read Oxford Nanopore sequencing on both Ludwigia peploides (Lpm) and Ludwigia grandiflora (Lgh)?
Answer: Lgh has a genome size of 1419 Mb, which is 5-fold larger than the Lpm genome (262 Mb). Therefore, we decided to maximize the sequencing coverage for Lgh by combining both SR and LR technologies.
Action: Sentences were added to the “library preparation and sequencing’ paragraph (lines 187-190)
- The phylogenetic methods should be described in more detail, including information on the
selected species, outgroup species, and the data used in phylogenetic analyses.
Action: The Materials and Methods section was modified in response to this comment (lines 254-263), and accession numbers were added to Figure 8 (phylogeny).
- Which DNA extraction kit was used?
Answer: Ludwigia species contain large amounts of polysaccharides and polyphenols, which interfere with the extraction of genomic DNA and make it difficult to obtain good quality DNA for sequencing. To extract DNA from Ludwigia sp, we first explored different standard DNA extraction kits, which failed to obtain good quality DNA. We then combined different purification steps using different types of Macherey-Nagel buffer, which resulted in DNA of a quality suitable for sequencing.
Action: We completed the sentence line 153-154
Lines 179, 190, 217: Please add a reference to all tools you used.
Action: The references have been added and all other references have been checked.
Line 191-192: Why does the number of ONT reads have a high difference between Lgh and
Lpm?
Answer: This is because, due to the genome size of Lgh, we performed 3 runs instead of just one for Lpm.
Action: This information has been added to the text at lines 187-190
Line 230: Ludwigia needs to be italicized. Species and genus names are sometimes given in
italics, sometimes not. Please check throughout the manuscript carefully.
Action: Done
Results
- Provide details about the quality of Illumina and Oxford Nanopore Sequencing reads,
including information on the quantity of data obtained, how much data was used for the
assembly, and its coverage.
Action: Number of reads were added lines 274 and 292 and coverage lines 288 and 298
Line 319: There is a mention to Table 2, but I think the authors meant Table 1.
Action: Thank you for pointing out this error, it has been corrected.
Line 315–319: In general variations results, it is better if also provide numerical information.
In general, providing numerical information would enhance the clarity of the variations
observed.???
Action: Numerical information were added lines 312-321
Line 346-347: In the results, the authors mentioned comparing Ludwigia sp. junctions with
those of other Onagraceae plastomes (Figure 5). The authors should provide details about
the analysis and the species used in the methods section.
Action: Details and accession numbers added lines 347-354 as well as in Figure 5 legend (lines 712-716)
In the phylogenetic tree, the authors only utilized matK sequences for constructing the tree.
Why did the authors not include additional gene or protein sequences to enhance the
phylogenetic analysis? Additionally, the authors should specify the source of the matK
sequences.
Answer: matK is the only protein-coding barcode available for the majority of Ludwigia species and was chosen based on this criterion.
Action: Accession numbers were added to Figure 8.
In the content, the authors mention "Add. Figure," but below the figure, the author used
"Supp. Figure.
Action: We homogenized using Add.
Discussion
It would be better if the authors discussed and expanded on how the identification of two
plastome haplotypes in Ludwigia grandiflora contributes to advancing phylogenetic and
evolutionary studies.
Answer: We have completed the tree, added Circaea as the outgroup, discussed the contribution of plastid markers and present the importance of expanding the complete plastome database of Ludwigia to advance phylogenetic and evolutionary studies.
Action: We add a new discussion paragraph lines 612-638 and change Figure 8 legend (lines 735-740)
, posted 28 May 2024, validated 28 May 2024Dear authors
First I ask for forgiveness for the delay, but it was complex to find suitable and willing reviewers. Two of them accepted to review your manuscript. As you will see in the reviews, while one is rather positive and asked for minor revisions, the second one asked for deeper ones.
After reviewing the paper myself, I think that the requests from both are legitimate, and propose you to answer them before acceptance
Sincelery yours
Francois Sabot
In this manuscript, the authors describe the chloroplast genome’s reconstruction of two species from Ludwigia genus. They used several approaches: short-read, long-read and long-read with correction using short-reads. They concluded that the hybrid method is the most efficient. Later, they described the composition of the genomes (number of genes, number and position of SSR, position of IR boundaries, etc.) and produced a phylogeny of the family based on one gene, matK.
The introduction is not very attractive. As the species is not well known, its description and a justification of its interest should be mentionned in the first paragraph.
The material and methods is written in a very confused way and it is frequently difficult to understand what the authors have done exactly.
For example, the authors said “Chloroplastic R1 and R2 reads were used with and without prior error correction using ONT reads with BayesHammer”. The sentence suggests that ONT reads where used to correct Illumina reads.
The whole process of genome reconstruction is unclear. As I understand, the authors reconstructed a first sequence using short-reads. Then they used this 1rst sequence to filter chloroplast reads and reconstruct four sequences using short-reads (using ABySS, MEGAHIT, Velvet and SPAdes ) and two sequences using long-reads (using FLYE and CANU). What’s the point of this two steps reconstruction? Why not using an existing whole chloroplast sequence to filter chloroplast reads then to reconstruct only two new sequences (one using short-reads, one using long-reads)?
I also wonder why presenting and comparing five different methods of reconstructions instead of the only one that was used to generate the sequences that areactually submitted to Genbank. In a technical paper talking about comparison of assembly software, I would understand, here I would rather read something more concise and focused.
The description of phylogeny reconstruction is confusing as well. “We reconstructed phylogenetic relationships among plastomes of Onagraceae. The FFT-NS-2 method in MAFFT 7 [55] was used to align all plastomes with one of the IRs removed to avoid data duplication.” suggests that the phylogeny was done using whole plastome sequences, while “we propose a phylogenetic tree from Ludwigia matK sequences (Figure 8).” suggests that the phylogeny was reconstructed using only one gene, more precisely only 149 amino acids.
The results and discussion are many times oversold.
“After conducting our research, we discovered that hybrid assembly, which incorporates both long and short read sequences, resulted in the most superior complete assemblies. This innovative approach capitalizes on the advantages of both sequencing technologies, harnessing the accuracy of short read sequences and the length of long read sequences.”. It seems that the authors discovered that hybrid assembly is more efficient. I’m surprised as this is well known (see for example Jain et al. Nature Biotech 2018 or Mak et al. MBE 2023) and already incorporated on chloroplast assembly pipeline (ex. ptGAUL, Zhou et al. Mol Ecol Res 2023).
Same comment about the flip-flop organization of the LSC. The existence of both haplotypes has already been described. This study adds one more species to the list of species where flip-flop organization was described, but it does not justify the paragraph in the discussion.
Similar comment about the evolution of IR. Despite being interesting, I’m not sure this discussion has its place on a paper announcing a new chloroplast sequence. This part deserves to be more developed and published alone, rather than buried in a technical paper.
Another important point is the absence of code lines used to analyse the data. Raw data (Illumina and ONT) are not provided. Without these information, the analysis is not reproducible.
Similarly, the authors did not mention the references of the samples used for the phylogeny.
Overall, I think that producing a new whole chloroplast sequence is important, especially for a poorly known species. However, such data should be published as a genome report, a technical paper, not as a research paper.
The manuscript entitled “Sequencing, de novo assembly of Ludwigia plastomes, and comparative analysis within the Onagraceae family” presents the first sequencing and assembly of complete plastomes for Ludwigia peploides (Lpm) and Ludwigia grandiflora (Lgh) using a hybrid assembly approach. It identifies two plastome haplotypes in Lgh and suggests the likely presence of two haplotypes in Lpm, expanding our understanding of plastome diversity within the Ludwigia genus.
It's an interesting manuscript that's well written and organized. However, there are a few things that, in my opinion, can be improved as mentioned below:
Evaluation of the various components of the article
Title and abstract
Does the title clearly reflect the content of the article? [X] Yes, [ ] No (please explain), [ ] I don't know
Does the abstract present the main findings of the study? [X] Yes, [ ] No (please explain), [ ] I don’t know
Introduction
Are the research questions/hypotheses/predictions clearly presented? [X] Yes, [ ] No (please explain), [ ] I don’t know
Does the introduction build on relevant research in the field? [X] Yes, [ ] No (please explain), [ ] I don’t know
· Materials and methods
Are the methods and analyses sufficiently detailed to allow replication by other researchers? [ ] Yes, [X] No (please explain), [ ] I don’t know
Some methods sections lack clear and detailed methods, such as phylogenetic tree building.
Are the methods and statistical analyses appropriate and well described? [ ] Yes, [X] No (please explain), [ ] I don’t know
The phylogenetic tree analyses need to be described in more detail.
· Results
In the case of negative results, is there a statistical power analysis (or an adequate Bayesian analysis or equivalence testing)? [ ] Yes, [ ] No (please explain), [X] I don’t know
No “negative” results.
Are the results described and interpreted correctly? [X] Yes, [ ] No (please explain), [ ] I don’t know
Discussion
Have the authors appropriately emphasized the strengths and limitations of their study/theory/methods/argument? [X] Yes, [ ] No (please explain), [ ] I don’t know
Are the conclusions adequately supported by the results (without overstating the implications of the findings)? [X] Yes, [ ] No (please explain), [ ] I don’t know
List of comments, questions and suggestions
Abstract
The abstract provides a concise overview of the study and key findings. However, it might be further improved by clarifying the importance of the study: Explain why these Ludwigia species are important. Provide which methods or specific sequencing technologies were used to generate the plastid genomes. Provide numerical information on the plastid genome size of three Ludwigia species.
Line 12-13: I suggest split this sentence in two: “The Onagraceae family, which belongs to the order Myrtales, consists of approximately 657 species and 17 genera, including the genus Ludwigia L., which is comprised of 82 species.”
Introduction
What is the importance of Ludwigia species? I suggest including 1-2 sentences in the introduction to highlight the importance of these species. Additionally, provide a more detailed rationale for the importance of studying plastid genomes in Ludwigia species.
Line 44, 107: Ludwigia needs to be italicized. Please check throughout the manuscript carefully.
Line 63: Add Reference.
Line 67: Add Reference.
Line 95-97: The sentence seems not complete “Recent long-read sequencing (> 1000 bp) provides strong evidence that terrestrial plant plastomes have two structural haplotypes, present in equal proportions and differing in IR orientation of the [22].”
Materials and methods
Why did authors not perform short-read Illumina sequencing and long-read Oxford Nanopore sequencing on both Ludwigia peploides (Lpm) and Ludwigia grandiflora (Lgh)?
The phylogenetic methods should be described in more detail, including information on the selected species, outgroup species, and the data used in phylogenetic analyses.
Which DNA extraction kit was used?
Line 179, 190, 217: Please add a reference to all tools you used.
Line 191-192: Why does the number of ONT reads have a high difference between Lgh and Lpm?
Line 230: Ludwigia needs to be italicized. Species and genus names are sometimes given in italics, sometimes not. Please check throughout the manuscript carefully.
Results
Provide details about the quality of Illumina and Oxford Nanopore Sequencing reads, including information on the quantity of data obtained, how much data was used for the assembly, and its coverage.
Line 319: There is a mention to Table 2, but I think the authors meant Table 1.
Line 315–319: In general variations results, it is better if also provide numerical information. In general, providing numerical information would enhance the clarity of the variations observed.
Line 346-347: In the results, the authors mentioned comparing Ludwigia sp. junctions with those of other Onagraceae plastomes (Figure 5). The authors should provide details about the analysis and the species used in the methods section.
In the phylogenetic tree, the authors only utilized matK sequences for constructing the tree. Why did the authors not include additional gene or protein sequences to enhance the phylogenetic analysis? Additionally, the authors should specify the source of the matK sequences.
In the content, the authors mention "Add. Figure," but below the figure, the author used "Supp. Figure.
Discussion
It would be better if the authors discussed and expanded on how the identification of two plastome haplotypes in Ludwigia grandiflora contributes to advancing phylogenetic and evolutionary studies.
Download the review