
Recommendation
 based on reviews by Gonzalo Riadi and 2 anonymous reviewers
based on reviews by Gonzalo Riadi and 2 anonymous reviewers
Whole genome sequences are revolutionizing our understanding across various biological fields. They not only shed light on the evolution of genetic material but also uncover the genetic basis of phenotypic diversity. The sequencing of underrepresented lineages, such as the one presented in this study, is of critical importance. It is crucial in filling significant gaps in our understanding of Metazoa evolution. Despite the wealth of genome sequences in public databases, it is crucial to acknowledge that some lineages across the Tree of Life are underrepresented or absent. This research represents a significant step towards addressing this imbalance, contributing to the collective knowledge of the global scientific community.
In this genome note, as part of the European Reference Genome Atlas pilot effort to generate reference genomes for European biodiversity (Mc Cartney et al. 2023), Klara Eleftheriadi and colleagues (Eleftheriadi et al. 2023) make a significant effort to add a genome sequence of an unrepresented group in the animal Tree of Life. More specifically, they present a taxonomic description and chromosome-level genome assembly of a newly described species of horsehair worm (Gordionus montsenyensis). Their sequence methodology gave rise to an assembly of 396 scaffolds totaling 288 Mb, with an N50 value of 64.4 Mb, where 97% of this assembly is grouped into five pseudochromosomes. The nuclear genome annotation predicted 10,320 protein-coding genes, and they also assembled the circular mitochondrial genome into a 15-kilobase sequence.
The selection of a species representing the phylum Nematomorpha, a group of parasitic organisms belonging to the Ecdysozoa lineage, is good, since today, there is only one publicly available genome for this animal phylum (Cunha et al. 2023). Interestingly, this article shows, among other things, that the species analyzed has lost ∼30% of the universal Metazoan genes. Efforts, like the one performed by Eleftheriadi and colleagues, are necessary to gain more insights, for example, on the evolution of this massive gene lost in this group of animals.
References
Cunha, T. J., de Medeiros, B. A. S, Lord, A., Sørensen, M. V., and Giribet, G. (2023). Rampant Loss of Universal Metazoan Genes Revealed by a Chromosome-Level Genome Assembly of the Parasitic Nematomorpha. Current Biology, 33 (16): 3514–21.e4. https://doi.org/10.1016/j.cub.2023.07.003
Eleftheriadi, K., Guiglielmoni, N., Salces-Ortiz, J., Vargas-Chavez, C., Martínez-Redondo, G. I., Gut, M., Flot, J.-F., Schmidt-Rhaesa, A., and Fernández, R. (2023). The Genome Sequence of the Montseny Horsehair worm, Gordionus montsenyensis sp. Nov., a Key Resource to Investigate Ecdysozoa Evolution. bioRxiv, ver. 3 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2023.06.26.546503
Mc Cartney, A. M., Formenti, G., Mouton, A., De Panis, D., Marins, L. S., Leitão, H. G., Diedericks, G., et al. (2023). The European Reference Genome Atlas: Piloting a Decentralised Approach to Equitable Biodiversity Genomics. bioRxiv. https://doi.org/10.1101/2023.09.25.559365
DOI or URL of the preprint: https://doi.org/10.1101/2023.06.26.546503
Version of the preprint: 1
, posted 18 Oct 2023, validated 18 Oct 2023Dear Rosa, I hope this email finds you well. First, I apologize for the long time the editorial process has taken. Getting people available to perform reviews takes a lot of work.
Your article has been reviewed, and there are some issues that need to be answered before we can recommend it. We are looking forward to the revised version of your article.
All the best
Juan C. Opazo
PCI Review
The genome sequence of the Montseny horsehair worm, Gordionus montsenyensis sp. nov., a key resource to investigate Ecdysozoa evolution
General questions
Did you read the “guide for reviewers”? (see the Help menu of the thematic PCI or the dedicated blog post)
R: yes.
Is the manuscript well written?
R: yes.
Is the description of the rationale and methods clear and comprehensive?
R: They are enough. Just a couple of points: the methodology of the tree building, and genome size estimation are lacking.
Are there flaws in the design of the research?
R: No.
Are there flaws in the analysis?
R: I missed the genome size estimation, but I wouldn’t qualify it as a major flaw. I suggest to add more information in the methodology and discussion of the manuscript, in the last section of comments of this review.
Are there flaws in the interpretation of results?
R: The Hi-C experiment suggests 5 chromosomes, but the assembly describes 6 main pseudochromosomes. This is not a flaw in the interpretation of results, but calls for a comment in the discussion.
Do you have concerns about ethics or scientific misconduct?
R: No.
Did you detect a spin on the results, discussion or abstract? (a spin is a way of twisting the reporting of results such that the true nature and range of the findings are not faithfully represented, https://doi.org/10.1073/pnas.1710755115)
R: No. The genome report, does not precise that the genome reported is partial. That would be desirable (maybe in the discussion as suggested in my comments, last section).
Is something critical missing?
R: Apart from the methodology of the tree and the genome size estimation, no. I suggest to add in the methodology and discussion about sequence alignment, in the last section of this review.
Evaluation of the various components of the article
Title/abstract/introduction
Does the title clearly reflect the content of the article?
R: I think the second phrase in the title, after the comma: “a key resource to investigate Ecdysozoa evolution” while being true, is not too well supported by the text, or this study.
Does the abstract present the supported findings of the study concerned and no other?
R: yes.
Does the introduction clearly explain the motivation for the study?
R: pretty much, yes.
Is the research question/hypothesis/prediction clearly presented?
R: The sequencing of the worm was justified.
Does the introduction build on relevant recent and past research performed in the field?
R: It builds on the necessary information about the biology of the worm in order to understand the need of its genome sequence. However, I would have liked to know a bit about what genomic information is currently available in the databases, if not from the particular species, from its relatives. (and a comparison of the numbers, in the discussion)
Materials and Methods
Are the methods and analysis described in sufficient detail to allow replication by other researchers?
R: Except for the tree, the aligment and the database used for Repeatmasker, yes.
Is the experimental plan consistent with the questions?
R: Generally, yes.
Are the statistical analyses appropriate?
R: Again, I missed the genome size estimation. The rest of the statistical analyses are standard and well performed by known available programs. I missed technical information on the sequence comparisons and the tree building, too.
Have you evaluated the statistical scripts and program codes?
R: No new scripts were developed in this work, so there was no need to evaluate scripts this time.
Results
Have you checked the raw data and their associated description?
R: The data was deposited in the European Nucleotide Archive, ENA, although it is not mentioned explicitly in the text. I also would have liked to see the Genome reports in Supplementary material, which contain more technical information about the sequencing, and genome or transcriptome sample quality checks. No information in the text, on where the genome assembly and annotation files are/will be available for download.
Have you run the data transformations and statistical analyses and checked that you get the same results?
R: No. However, the results are sound respect to the raw data information deposited online, methodology described, and from the tables and figures provided. Discrepancies (like chromosome number, completeness of the assembly, number of protein coding genes, comparisons with other relatives), however, should be discussed in the appropriate section, but are not.
To the best of your ability, can you detect any obvious manipulation of data (e.g. removal)?
R: No.
Do the statistical results strongly support the conclusion (p< 10-3 or BF>20)?
R: Although no hypothesis testing was explicitly performed, so no p-value of Bayes Factors were calculated, genome data analysis was done correctly.
In the case of negative results, was a statistical power analysis (or an appropriate Bayesian analysis) performed?
R: No “negative” results as in “hypothesis disproved” in a genome report. In the Discussion section, though, a comment on how complete the genome was sequenced and assembled; and a comment on the final number of chromosomes (why they have 6 pseudochromosomes when Hi-C suggests 5) would be desirable.
Did the authors conduct many experiments but retain only some of the results?
R: No.
Discussion
Do the interpretations of the analysis go too far?
R: No.
Are the conclusions adequately supported by the results?
R: Yes.
Does the discussion take into account relevant recent and past research performed in the field?
R: The discussion is centered in the worm’s biology, not its genome sequence, assembly or annotation. It would be interesting to read about the comparison of the manuscript genome assembly results, like genome size and number of protein coding genes, with relative species.
Did the authors test many hypotheses but consider only a few in the discussion?
R: No. This is a genome report, describing the sequencing, assembly and annotation of a genome, not a research article testing hypotheses.
References
Are all the references appropriate?
R: Yes.
Are the necessary references present?
R: Yes.
Do the references seem accurate? R: Yes.
Tables and figures
Are the tables and figures clear and comprehensive?
R: Yes.
Are all the tables/figures useful?
R: No.
Are there too many/too few tables and figures?
R: Yes. Figure 5, Figure 7 and Figure 8 could go in supplementary material.
Do the tables and figures have suitable captions such that they can be understood without having to read the main text?
R: No. Figure 2. Please, enrich either the caption or the methodology with more information. For instance, what features were used for generating the tree? What biological sequences? The list of accessions should be available in the supplementary materials. How were they processed? What are the outgroups?
Figure 4 could be further explained. The diploidy peak, and the repeats peak, for example. Is the x-axis k-mer coverage or genome coverage?
Comments, questions and suggestions
Abstract suggestions
Change “the most neglected” for “one of the less studied”. Neglected is an active verb, with an emotional load, whereas “less studied” is passive, and probably corresponding to what actually happens.
Introduction suggestions
Add a comma after, “As expected” in the first paragraph.
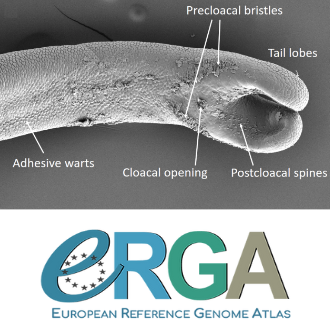
Figure 1. The electron micrography shows a male. How come it was not described in the materials examined?
Genome Sequence Report
“340x coverage of long reads and 80x coverage of small reads”. This is respect to which genome size? One genome previously reported? Maybe one from a cytogenetic study? Or from a genome size estimation? Or from the final assembled genome size? Genome size estimation (and final coverage) is important, since it can be compared with the initial coverage and the assembled size and to estimate how much sequencing was “wasted”, and how much genome is left to be sequenced. Also, the coverage has to be specified, initial (previous analysis, just after sequencing) or final (after analysis, quality control, trimming and genome size estimation, previous assembly). FASTQC files should go in the supplementary materials.
“Pair-wise (sequence) similarity is 95.04%” (page 3, just before Discussion). Sequence similarity or sequence identity? Please specify. How was the alignment done (global, local, algorithm, parameters)? Were mitochondrial genomes used as pointed out two paragraphs later, in Discussion section? DNA or protein sequences? This, I believe, was not specified in the Methodology.
“warty appearance of spines of G. montsenyensis is unique and justifies the description as a new species.” I am not an expert, so I do not feel knowledgeable about the criteria to consider a particular worm as new species. However, as “Nematomorphs are not rich in characters that can be used for identification”, a study using biological sequences (particularly the ITS region known to be important for species determination in worms), both only sequence or phylogenetic could enlighten this question as supporting information. Discussion is lacking in this respect, connecting the phylogenetic analysis with the idea that this is a new species.
“96% of the assembly sequence assigned to 6 pseudochromosomes”. However, Figure 6 suggests only 5 chromosomes. Please, comment.
Figure 2. Please, enrich either the caption or the methodology with more information. For instance, what features were used for the tree building? What biological sequences? DNA or proteins? The list of accessions should be available in the supplementary materials. How were they processed? What are the outgroups? The final log should go in the supplementary materials.
Figure 4 could be further explained. The diploidy peak, and the repeats peak, for example. Is the x-axis k-mer coverage or genome coverage?
Figure 5, Figure 7 and Figure 8 could go in supplementary material.
Figure 7 has y-axis title base outward respect to the axis. Please, rotate the y-title 180 degrees so the title reads from bottom up.
How many reads were sequenced for the transcriptome? This is not reported in the methodology. Do the authors think that, in spite of 96% of genome sequence assembled, lack of an estimated 40% of genes by BUSCO in the final genome could be, at least in part, due to the tissue sampling, or not enough depth in transcriptome sequencing? OR could it be due to an assembly problem? A comment about this could enrich the Discussion section.
Please, specify and reference the database that was used together with Repeatmasker in Repeat Identification section.
Latin expressions like “de novo” or “ab initio” should go in italics, in the text.
No information from were to download the genome assembly and annotation in the text.
https://doi.org/10.24072/pci.genomics.100252.rev11In writing this review, I firstly want to flag potential conflicts of interest. I am myself a member of the large team working on the ERGA pilot, I am also collaborating with some of the authors on projects outside of ERGA, and I am host to one of the authors on their MSCA project. I assume that for genome reports as this one, such situations cannot be avoided, simply due to the extremely high number of reports that will be written as part of the EBP.
In their report on the genome of a newly discovered nematomorph species, Gordionus montsenyensis sp. nov., the authors formally describe the species, and describe the genome. The report is very well written and includes all necessary information to be published. I have two suggestions:
1. The authors state, "None of these sequences were filtered, since due to the parasitic life cycle of nematomorphs, these sequences could be horizontally transferred sequences." They could easily include a screen for potential HGT candidates into their report.
2. The authors split their report into a section that uses classical morphology to describe the species, and then a second one to describe the genome. I think the morphological description is very valuable, but would still want to suggest for the authors to incorporate the genome into the species description. That is, used basic features of the genome as additional information to describe the new species.
Additional minor points:
"In this piece of work," -> please consider to use different wording
"this enigmatic animal phyla" -> phylum
"As expected given their parasitic lifestyle,"" -> this needs a citation if it is really expected
https://doi.org/10.24072/pci.genomics.100252.rev12This study introduces a newly discovered species of Nematomorpha, Gordionus montsenyensis Schmidt-Rhaesa & Fernández sp. nov., along with a chromosome-level genome assembly. The assembly comprises 398 scaffolds, totaling 288 Mb, and boasts an impressive N50 of 52.6 Mb, with 96% of the genome organized into 6 pseudochromosomes. Additionally, the authors have successfully assembled a 15-kilobase circular mitochondrial genome and identified 10,819 protein-coding genes. This valuable genomic resource significantly contributes to the exploration of Ecdysozoa evolution and enhances our understanding of the genetic foundations of parasitic lifestyles.
Point of concern:
1. The author focuses on N50, it is a metric that provides insight into the 'average' sizes of long scaffolds within an assembly. This measurement is particularly effective when the genome assembly is "predominantly error-free".
I suggest the author assess assembly quality by examining the k-mer distribution in the assembly and compare it to the expected k-mer distribution derived from the sequencing reads. By utilizing this k-mer correctness metric in conjunction with N50, we can gain insight into whether a high N50 value is the result of mostly accurate junctions or if it is influenced by numerous incorrect junctions.
2.The English in some sections of the manuscript could benefit from minor improvements. For instance, the statement "The nematomorph fauna from Spain is only fragmentarily known" could be rephrased as "Our understanding of the nematomorph fauna in Spain is limited and fragmented" to enhance clarity.
3. Maximum likelihood phylogenetic tree “Figure 2” illustrating the positioning of Gordionus montsenyensis sp., with support values of 83/85/85 for standard nonparametric bootstrap, SH-aLRT and UFBoot.
Generally, we tend to place more confidence in a clade when its SH-aLRT exceeds 80% and UFBoot exceeds 95%. I would recommend providing the Average Nucleotide Identity (ANI) values to support this relationship.
4. Page number 14 “The size of each circle is proportional to the scaffold length and each color represents taxonomic assignment by a blast search against the nt database”
Did you take the second-best match into account during your assignment? It's recommended to employ the Kraken k-mer-based approach and conduct a cross-comparison of the results for enhanced accuracy.
5. On page 17, could you elaborate on your rationale for selecting 'with k=27' for the analysis? Please provide an explanation for this choice.
6. On page 18, “The mitochondrial genome was assembled with MITGARD (Nachtigall, Grazziotin, and Junqueira-de-Azevedo 2021) using the WGS Illumina paired-end reads and the mitochondria of Gordionus alpestris (NC_044095.1) as a reference and selecting the clade Arthropoda. The mitochondrial genome was annotated using MitoZ with parameters annotate--genetic_code auto--clade Arthropoda (Meng et al. 2019)”
MITGARD is specifically crafted to extract the mitochondrial genome from "RNA-seq" data of various Eukaryotic species. To clarify the type of data involved in mitochondrial genome assembly, it's important to note that MITGARD focuses on RNA-seq data, which differs from approaches like MitoFinder, which exclusively rely on whole genome sequencing reads for assembly. It is also designed to find and annotate mitochondrial sequences in existing genomic assemblies. To further illustrate the contrast, it would be valuable to include comparative results between MITGARD and MitoFinder, and explain the methods in detail for clarity.
7. Table 1 highlights the completeness of BUSCO*completeness C:59.5%[S:59.3%,D:0.2%],F:12.1%,M:28.4:%,n:954. It suggests a relatively low completeness, and a lots of missing fragments which may indirectly imply potential issues with the assembly. It would be highly valuable to include a KAT kmer completeness score for a more comprehensive evaluation of the data quality.
https://doi.org/10.24072/pci.genomics.100252.rev13
