
Recommendation
 based on reviews by Maria Recuerda and 1 anonymous reviewer
based on reviews by Maria Recuerda and 1 anonymous reviewer
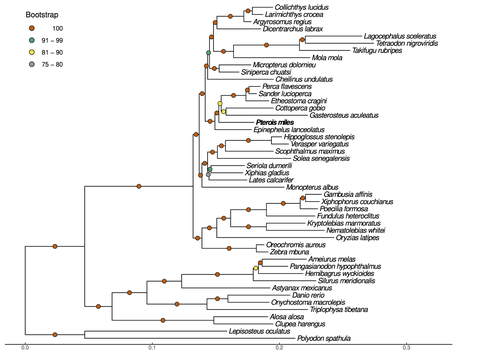
High-quality genomes are currently being generated at an unprecedented speed powered by long-read sequencing technologies. However, sequencing effort is concentrated unequally across the tree of life and several key evolutionary and ecological groups remain largely unexplored. So is the case for fish species of the family Scorpaenidae (Perciformes). Kitsoulis et al. present the genome of the devil firefish, Pterois miles (1). Following current best practices, the assembly relies largely on Oxford Nanopore long reads, aided by Illumina short reads for polishing to increase the per-base accuracy. PacBio’s IsoSeq was used to sequence RNA from a variety of tissues as direct evidence for annotating genes. The reconstructed genome is 902 Mb in size and has high contiguity (N50=14.5 Mb; 660 scaffolds, 90% of the genome covered by the 83 longest scaffolds) and completeness (98% BUSCO completeness). The new genome is used to assess the phylogenetic position of P. miles, explore gene synteny against zebrafish, look at orthogroup expansion and contraction patterns in Perciformes, as well as to investigate the evolution of toxins in scorpaenid fish (2). In addition to its value for better understanding the evolution of scorpaenid and teleost fishes, this new genome is also an important resource for monitoring its invasiveness through the Mediterranean Sea (3) and the Atlantic Ocean, in the latter case forming the invasive lionfish complex with P. volitans (4).
REFERENCES
1. Kitsoulis CV, Papadogiannis V, Kristoffersen JB, Kaitetzidou E, Sterioti E, Tsigenopoulos CS, Manousaki T. (2023) Near-chromosome level genome assembly of devil firefish, Pterois miles. BioRxiv, ver. 6 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2023.01.10.523469
2. Kiriake A, Shiomi K. (2011) Some properties and cDNA cloning of proteinaceous toxins from two species of lionfish (Pterois antennata and Pterois volitans). Toxicon, 58(6-7):494–501. https://doi.org/10.1016/j.toxicon.2011.08.010
3. Katsanevakis S, et al. (2020) Un- published Mediterranean records of marine alien and cryptogenic species. BioInvasions Records, 9:165–182. https://doi.org/10.3391/bir.2020.9.2.01
4. Lyons TJ, Tuckett QM, Hill JE. (2019) Data quality and quantity for invasive species: A case study of the lionfishes. Fish and Fisheries, 20:748–759. https://doi.org/10.1111/faf.12374
DOI or URL of the preprint: https://doi.org/10.1101/2023.01.10.523469
Version of the preprint: 5
Dear recommender,
We are deeply grateful for your comments and corrections. We have made all suggested corrections and read through the manuscript again to identify possible remaining mistakes. We now believe that it is ready for recommendation.
With best regards,
Tereza Manousaki (on behalf of all authors)
, posted 16 May 2023, validated 16 May 2023Dear authors,
Thank you very much for submitting your revised manuscript. I think it has significantly improved after incorporating the reviewers’ comments. I will be delighted to recommend your work.
I found some minor typos, listed below. I think this is a good moment to re-read the manuscript and fix small details before the work is published.
L25: in the species’ biology and ecology
L50: considered to have a major
L53: invasion dynamics
L156: from contig sizes
L181: A step of header correction
L226: check “non-redundant over”, is this correct?
L235: Were aligned to the genome assembly
L237: Ab initio prediction on the P. miles
L248: that overlapped with TEs
L286: What does “higher than scaffold level” mean?
L303: were concatenated into a superalignment
L340: Duplication event estimation
L361: Toxin gene evolution in lionfishes
L436: lowercase “phylogenomics”
L437: The total number of genes or proteins? >1 Mio. genes seems unlikely.
L444. 100% non-parametric bootstrap. Please also check to specify throughout that this is non-parametric bootstrapping (e.g., Figures 2, 6). Not to be confused with parametric bootstrapping.
L475: please, provide means and standard deviations
L553: it could be worth
L1181: Percentage of transposable element category representation
L1222: Number of orthogroups associated (gene families ≠ Orthofinder’s orthogroups)
L1246: Transposable element annotation statistics
DOI or URL of the preprint: https://doi.org/10.1101/2023.01.10.523469
Version of the preprint: 3
Dear editor,
We thank you deeply for the comments and the time and insights offered by all three of you that have greatly improved our manuscript. We have addressed the raised points and respond to them one-by-one below, hoping that our replies will render the manuscript appropriate for recommendation.
Sincerely,
Tereza Manousaki, on behalf of all authors
One-be-one responses to the comments
I think the introduction could make a better case for usefulness of this new genome. For example, as resource for monitoring invasion dynamics with population genomics, or perhaps to study the hybrid origin of Pterois species complex in the Atlantic.
>>Thank you for the proposal. The following text has been added to the introduction of the revised manuscript:
“Data derived from Whole Genome Sequencing (WGS) could provide promising opportunities in the exploration of potential adaptations that shape the fitness of invaders, as well as the dynamics of colonization. Further, it will provide a basis for understanding the hybrid origin of the invasive lionfishes (P. miles and P. volitans) in the Western Atlantic (Wilcox et al., 2017).”
Mitogenome. It would be ideal if the mitogenome of this specimen is also assembled from the raw data and compared to the available sequences by Dray et al. 2016.
>This was indeed a very interesting suggestion. Unfortunately, we do not have the mitogenome in our assembled contigs. To explore the reason for that, we did blast searches at the raw data, and only few sequences including mitochondrial genes were present. For that reason, we could not move forward with identifying and describing the mitogenome.
Karyotype. Is there any information on the karyotype of this species or a close relative? I believe this is an important point to talk about near-chromosome-scale assemblies, but also very interesting from an evolutionary viewpoint.
>>We know the karyotype of the sister species Pterois volitans (Nirchio et al., 2014) and have added this information in the text, as follows:
“Taking into consideration that the haploid number of P. miles should be the same as its con-generic species, P. volitans, n=24 (Nirchio et al., 2014),”
Reviewer 2 suggests some additional analyses. In addition, I think it would be worth exploring the genes that are responsible for its venomousness, which is a very special characteristic of the species.
>> This has been a major addition to this manuscript. Thank you for the suggestion. We have identified the toxin-producing genes in P. miles and presented their phylogenetic relationship with other genes within the family.
Transposable elements. Reviewer 2 also suggests to further explore the reasons for P. miles’ high TE proportion. I agree that this is a very interesting topic, but also potentially complex given that TE landscapes can also vary due to the use of different bioinformatic pipelines in their identification. Taking into account this, would the higher TE proportion identified in P. miles be likely a methodological or biological difference or both? For a biological explanation, it could be that the higher TE contents of P. miles make the genome more dynamic and thus more able to rapidly adapt to changing conditions, which could partly explain its success in invading new environments. For example, a similar hypothesis has been proposed for Mytilus muscles (https://genomebiology.biomedcentral.com/articles/10.1186/s13059-020-02180-3). Could a similar situation also occur in P. miles?
>> Thank you for this comment. We have been also considering the reason for the high TE content in P. miles, but still a clear conclusion could not be drawn. We have expanded this topic in the discussion to transfer the main hypotheses to the reader, which are the potential role in the success in the invasion as proposed also by the reviewer, potential methodological issues, or other unrecognizable yet reasons. However, only a thorough and taxonomically wide survey of TEs using the same methodology across fish would provide evidence for understanding deeper how TEs may be linked to invasiveness in fish.
The respective text reads:
“All aforementioned TE analyses in the different genomes, including ours presented herein, have been implemented using a different strategy for identifying the TEs of each species, a fact that could bias the results not allowing us to do a direct comparison. However, P. miles TE content exceeds remarkably most other fish genomes, with potential biological role in the species evolution. Elevated genome-wide repeat content has been previously linked to adaptation (Yuan et al., 2018) and invasiveness (Stapley et al., 2015; Danis et al., 2020) Thus, this higher proportion of TEs in the genome of P. miles could potentially play a key factor in adaptive evolution of species and consequently success in thriving in new environments.”
Synteny analyses. We see that synteny is generally conserved to G. aculeatus, but is this expected in actinopterygian fishes or exceptional? I would think that some interesting patterns could be identified (e.g., major fusions or fissions?) from this data.
>> Thank you for the comment! Even though stickleback is a model fish species, it has fewer chromosomes compared to most other teleosts, rendering it a rather suboptimal reference for finding fusions and fissions on our 24-chromosome focal species. This, combined with the fact that our assembly was not chromosome level, made us hesitate to deepen our synteny analysis as proposed by the reviewer. We hope that when we will have a chromosome level assembly, we will be able to perform the suggested analyses.
Orthogroup expansion and contraction. OG and gene families are treated as synonymous, but this is problematic. OGs try to approximate gene families but it is well known that automatic clustering approaches like Orthofinder has serious limitations, for example, for fast-evolving gene families being split into multiple OGs (e.g., Natsidis et al. 2021 10.1016/j.isci.2021.102110).
>> We have now clarified that with gene families we refer to orthogroups, making sure that the readers are not confused. See the text:
“Then, these GO terms and their descriptions were grouped/mapped into the gene families of their genes (HOGs from OrthoFinder), which were previously identified as rapidly expanding from CAFE and involved in duplication events by GeneRax.”
Also, please add how many HOGs were initially and after filtering.
>>This is included in the manuscript, see text:
“The total number of genes included in the proteomes of all 47 teleost fish species (Supplementary Table 1) and analyzed by OrthoFinder, was 1,108,753 and 97.8% of them were assigned to 28,397 phylogenetic HOGS. After the filtering step, 1,193 HOGs were selected to construct the superalignment matrix."
OG expansion/contraction analyses are presented from the whole tree, but the specific OGs that expand/contract in P. miles are not explored and this could provide interesting information about the species biology (e.g. venomousness).
>> Thank you! Venomousness is now explicitly studied in the new version of the manuscript.
Line 132 mentions draft, intermediate, and final assemblies. It would be good to make explicit which assemblies this refers to.
>>We have now rephrased the text describing the assembly process, and hope that it is now clearer.
Line 239. Note that BLAST’s “-max_target_seqs 1” does not warrant that the obtained hit is the best one (see https://doi.org/10.1093/bioinformatics/bty833)
>> We totally agree on this, however, given the use of a reciprocal best hit approach, we believe that the final outcome would not include extra false positives due to that parameter.
Line 278. What do you mean “in case of missing taxa, the aligned orthogroups sequences were corrected”? Do gaps or Ns not account for this directly, or is there anything I don’t get?
>>This was written inappropriately, and we thank you for pointing it out. We have rephrased and hope that now what we have done is explained clearly.
The manuscript reads very well, but some small English issues remain, please double check: missing “the” (line 55), weird phrasing in line 69, “library” (line 73), “Pfam domains were obtained” (line 249), “at the gene level” (line 291), “family” (line 299), “high confidence” (line 362), “the P. miles genome” (line 440), “TE superfamilies” (line 453), check also lines 263, 322, 324, 356.
Acknowledgements. Do we want to acknowledge the scuba diver that provided the sample with her/his name?
>>This has been one of the many scuba divers that collaborate with Cretaquarium for providing the exhibition with animals. We do not really have their name, but we have acknowledged the aquarium team that provided us with one of the lionfishes instead of adding them to their exhibition.
Reviewer 1:
Kitsoulis and collaborators provide the assembly and annotation of the devil firefish genome at a near-chromosome level, being the first available assembly of the family Scorpaenidae. The importance of this valuable reference genome is highlighted due to the invasive nature of the species, and therefore the need to generate the reference genome that provides the missing resource to study the biology, ecology and phylogeny of this successful invader.
The methodology is adequate for the main goal of the study. The combination of long-read MinION sequencing with more accurate Illumina short reads is a good approach to obtain a high-quality and complete genome assembly, as confirmed by the statistics reported in the article, including the N50, the BUSCO score and the circos plot showing the synteny with the 21 chromosomes of G. aculeatus . The maximum likelihood phylogenetic tree shows highly supported nodes placing the devil firefish within the Perciformes clade, being the first using genomic data. Regarding the genome structural and functional annotation, the implementation of three approaches yielded a very complete set of gene models. The characterization of TEs and gene losses and duplications is also very complete, providing a valuable resource for studying the role of these genomic variations in evolution and the adaptive potential of an invasive species like the devil firefish.
Overall, I consider that the study is very interesting and the methodology implemented is correct and matches the goals of the research. Therefore, as far as I am concerned, I find this article to be scientifically significant and technically accurate. I would just like to propose a few minor changes/recommendations to facilitate the reading.
>> We thank the reviewer deeply for the comments and the nice words.
I would mention the Tables and figures in order. For instance, the first table and figure that are mentioned are table and figure 3. I would reorder the tables to match the appearance in the text.
>>Done, thank you!
In order to reduce the number of figures, I would consider moving Figure 3 to supplementary materials and including the L50 in Table 3 (that is I mentioned in the previous comment it should be table 1).
>>Done, thank you!
Reviewer 2:
Kitsoulis and colleagues sequenced, assembled and annotated the genome of the devil firefish, Pterois miles, a fish species that has successfully invaded several non-native habitats (including the Mediterranean Sea from the Red Sea). They then deeply characterized the firefish transposable element (TE) landscape and, by including it in a phylogenetic framework with >40 other fishes, carried out a series of comparative genomic analyses in order to investigate the occurrence of similarities/differences between different genomic features in P. miles.
This paper is very well written and easy to follow, the analyses performed are robust, extensively explained and rich of methodological details. The programs and codes used, both available and devised appositely for this study, are appropriately described along the text and provided in publicly accessible repositories.
The genome assembly statistics are good, with excellent levels of completeness and good contiguity metrics. Overall, this is a rigorous study reporting the genome assembly of an ecologically key species, especially in the light of global warming that will further favor its success as invasive species. Such high-quality genome will help future studies that will rely, for example, on population-based individual resequencing to investigate the genomic consequences of such biological invasions.
>> Thank you for pointing out in such a clear way the importance of this work. We hope that it will help downstream research on understanding the population and invasion genomics of this successful invader.
That said, I only have a few minor comments that I hope will help to improve this study:
1) Is anything known about the karyotype of this species? If the karyotype is known, it could be interesting to compare the number of near-chromosomes assembled with the real number of chromosomes to have another estimate of the quality (contiguity in this case) of the assembly.
>>Thank you, see comment above.
2) On the same lines, I see that the authors, when describing the pipeline used for the genome assembly with the software Flye, mentioned a “genome size estimation of 900Mb”. Did the author estimate genome size in this study? This can be quickly done with a Kmer-based approach (e.g., with GenomeScope) using the Illumina reads, and might represent a nice addition to this study.
>>We know the genome size of the sister species P. volitans and used this as a genome size estimation of 900Mb. On top of that, we obtained a reference genome of 902.3 Mb from our assembly pipeline. Finally, our Illumina data were too few to run a k-mer analyses. We tried to do that but we did not get any meaningful results.
3) The authors carried out a very rigorous analysis to characterize the TE landscape of this fish species, and I very much like the approach they used. It is striking to see that TEs make a large part of the genome (>46%), more than usually seen in fishes with similar genome size. I am sure it is quite difficult/impossible to interpret the functional meaning of such TE expansion in this lineage, but I would like to see something about the expansion history of these TEs. It would be interesting to check whether such expansion was old or recent, and which TE classes expanded in certain times. Kimura substitution levels could be used to calculate and then draw the TE history.
>> Thank you for this point. We believe that this analysis, would be essential in a comparative mode, with other teleosts ideally also other members of the family Scorpaenidae, to understand the evolution of TEs especially coupled with the invasiveness success of various species. However, as this manuscript focuses on the overall genome analysis of P. miles, we would consider that it goes beyond the scope of our current paper.
4) Why do not perform a positive selection analysis on the whole gene set? The authors generated all the required input files and the appropriate phylogenetic framework, the same used for the gene contraction/expansion analysis. With some little more work one could investigate positive selection acting on the genes in the firefish lineage, and then making gene function analysis (e.g., GO terms) on the candidate genes found to be under positive selection. This could be discussed in the light of the lineage-specific/unique phenotypes of this species (for example the spines).
>> This is indeed another interesting analysis. We would prefer to implement this, when other genomes of the family are available as well, to be able to retrieve more specific target genes. We thank the reviewer for the suggestion, and will definitely implement this in a future work with more genomes hopefully soon.
5) I would make Fig. 1 and Fig. 2 supplementary material, there is not critical information for the main message of this study here, only technical details in the former, little information than can be included in the text in the latter. Another nice main figure, possibly included in a muti-panel figure with the current Fig. 4, can be the TE history plot (see my comment nr. 3 above).
>>To reduce the number of figures we have actually moved to the Supplements Figure 3 as suggested by Reviewer 1 as the two first figures describe the pipelines that we have developed and provide in the codes produced here, so hopefully they could be of use.
6) “Table 7. Completeness assessment (%)…”: Is this a BUSCO completeness assessment? I would make it clear in the table legend.
>> It has been added, thank you.
, posted 06 Mar 2023, validated 07 Mar 2023Dear authors,
Thank you very much for submitting your preprint to PCI Genomics. I received comments from two reviewers (see below) that found your work of very high quality. In addition, I have reviewed the preprint myself and arrived to similar conclusions. Below I provide some comments that I hope could be useful to improve the manuscript. Once my comments and those of the two reviews are addressed, I will be more than happy to recommend this work. Note that addressing the comments does not necessarily mean performing all the analyses, but I would require a point-by-point response letter.
I think the introduction could make a better case for usefulness of this new genome. For example, as resource for monitoring invasion dynamics with population genomics, or perhaps to study the hybrid origin of Pterois species complex in the Atlantic.
Mitogenome. It would be ideal if the mitogenome of this specimen is also assembled from the raw data and compared to the available sequences by Dray et al. 2016.
Karyophyte. Is there any information on the karyotype of this species or a close relative? I believe this is an important point to talk about near-chromosome-scale assemblies, but also very interesting from an evolutionary viewpoint.
Reviewer 2 suggests some additional analyses. In addition, I think it would be worth exploring the genes that are responsible for its venomousness, which is a very special characteristic of the species.
Transposable elements. Reviewer 2 also suggests to further explore the reasons for P. miles’ high TE proportion. I agree that this is a very interesting topic, but also potentially complex given that TE landscapes can also vary due to the use of different bioinformatic pipelines in their identification. Taking into account this, would the higher TE proportion identified in P. miles be likely a methodological or biological difference or both? For a biological explanation, it could be that the higher TE contents of P. miles make the genome more dynamic and thus more able to rapidly adapt to changing conditions, which could partly explain its success in invading new environments. For example, a similar hypothesis has been proposed for Mytilus muscles (https://genomebiology.biomedcentral.com/articles/10.1186/s13059-020-02180-3). Could a similar situation also occur in P. miles?
Synteny analyses. We see that synteny is generally conserved to G. aculeatus, but is this expected in actinopterygian fishes or exceptional? I would think that some interesting patterns could be identified (e.g., major fusions or fissions?) from this data.
Orthogroup expansion and contraction. OG and gene families are treated as synonymous, but this is problematic. OGs try to approximate gene families but it is well known that automatic clustering approaches like Orthofinder has serious limitations, for example, for fast-evolving gene families being split into multiple OGs (e.g., Natsidis et al. 2021 10.1016/j.isci.2021.102110). Also, please add how many HOGs were initially and after filtering.
OG expansion/contraction analyses are presented from the whole tree, but the specific OGs that expand/contract in P. miles are not explored and this could provide interesting information about the species biology (e.g. venomousness).
Line 132 mentions draft, intermediate, and final assemblies. It would be good to make explicit which assemblies this refers to.
Line 239. Note that BLAST’s “-max_target_seqs 1” does not warrant that the obtained hit is the best one (see https://doi.org/10.1093/bioinformatics/bty833)
Line 278. What do you mean “in case of missing taxa, the aligned orthogroups sequences were corrected”? Do gaps or Ns not account for this directly, or is there anything I don’t get?
The manuscript reads very well, but some small English issues remain, please double check: missing “the” (line 55), weird phrasing in line 69, “library” (line 73), “Pfam domains were obtained” (line 249), “at the gene level” (line 291), “family” (line 299), “high confidence” (line 362), “the P. miles genome” (line 440), “TE superfamilies” (line 453), check also lines 263, 322, 324, 356.
Acknowledgements. Do we want to acknowledge the scuba diver that provided the sample with her/his name?
Reviewer 1:
Kitsoulis and collaborators provide the assembly and annotation of the devil firefish genome at a near-chromosome level, being the first available assembly of the family Scorpaenidae. The importance of this valuable reference genome is highlighted due to the invasive nature of the species, and therefore the need to generate the reference genome that provides the missing resource to study the biology, ecology and phylogeny of this successful invader.
The methodology is adequate for the main goal of the study. The combination of long-read MinION sequencing with more accurate Illumina short reads is a good approach to obtain a high-quality and complete genome assembly, as confirmed by the statistics reported in the article, including the N50, the BUSCO score and the circos plot showing the synteny with the 21 chromosomes of G. aculeatus . The maximum likelihood phylogenetic tree shows highly supported nodes placing the devil firefish within the Perciformes clade, being the first using genomic data. Regarding the genome structural and functional annotation, the implementation of three approaches yielded a very complete set of gene models. The characterization of TEs and gene losses and duplications is also very complete, providing a valuable resource for studying the role of these genomic variations in evolution and the adaptive potential of an invasive species like the devil firefish.
Overall, I consider that the study is very interesting and the methodology implemented is correct and matches the goals of the research. Therefore, as far as I am concerned, I find this article to be scientifically significant and technically accurate. I would just like to propose a few minor changes/recommendations to facilitate the reading.
I would mention the Tables and figures in order. For instance, the first table and figure that are mentioned are table and figure 3. I would reorder the tables to match the appearance in the text.
In order to reduce the number of figures, I would consider moving Figure 3 to supplementary materials and including the L50 in Table 3 (that is I mentioned in the previous comment it should be table 1).
Reviewer 2:
Kitsoulis and colleagues sequenced, assembled and annotated the genome of the of devil firefish, Pterois miles, a fish species that has successfully invaded several non-native habitats (including the Mediterranean Sea from the Red Sea). They then deeply characterized the firefish transposable element (TE) landscape and, by including it in a phylogenetic framework with >40 other fishes, carried out a series of comparative genomic analyses in order to investigate the occurrence of similarities/differences between different genomic features in P. miles.
This paper is very well written and easy to follow, the analyses performed are robust, extensively explained and rich of methodological details. The programs and codes used, both available and devised appositely for this study, are appropriately described along the text and provided in publicly accessible repositories.
The genome assembly statistics are good, with excellent levels of completeness and good contiguity metrics. Overall, this is a rigorous study reporting the genome assembly of an ecologically key species, especially in the light of global warming that will further favor its success as invasive species. Such high-quality genome will help future studies that will rely, for example, on population-based individual resequencing to investigate the genomic consequences of such biological invasions.
That said, I only have a few minor comments that I hope will help to improve this study:
1) Is anything known about the karyotype of this species? If the karyotype is known, it could be interesting to compare the number of near-chromosomes assembled with the real number of chromosomes to have another estimate of the quality (contiguity in this case) of the assembly.
2) On the same lines, I see that the authors, when describing the pipeline used for the genome assembly with the software Flye, mentioned a “genome size estimation of 900Mb”. Did the author estimate genome size in this study? This can be quickly done with a Kmer-based approach (e.g., with GenomeScope) using the Illumina reads, and might represent a nice addition to this study.
3) The authors carried out a very rigorous analysis to characterize the TE landscape of this fish species, and I very much like the approach they used. It is striking to see that TEs make a large part of the genome (>46%), more than usually seen in fishes with similar genome size. I am sure it is quite difficult/impossible to interpret the functional meaning of such TE expansion in this lineage, but I would like to see something about the expansion history of these TEs. It would be interesting to check whether such expansion was old or recent, and which TE classes expanded in certain times. Kimura substitution levels could be used to calculate and then draw the TE history.
4) Why do not perform a positive selection analysis on the whole gene set? The authors generated all the required input files and the appropriate phylogenetic framework, the same used for the gene contraction/expansion analysis. With some little more work one could investigate positive selection acting on the genes in the firefish lineage, and then making gene function analysis (e.g., GO terms) on the candidate genes found to be under positive selection. This could be discussed in the light of the lineage-specific/unique phenotypes of this species (for example the spines).
5) I would make Fig. 1 and Fig. 2 supplementary material, there is not critical information for the main message of this study here, only technical details in the former, little information than can be included in the text in the latter. Another nice main figure, possibly included in a muti-panel figure with the current Fig. 4, can be the TE history plot (see my comment nr. 3 above).
6) “Table 7. Completeness assessment (%)…”: Is this a BUSCO completeness assessment? I would make it clear in the table legend.
Kitsoulis and colleagues sequenced, assembled and annotated the genome of the of devil firefish, Pterois miles, a fish species that has successfully invaded several non-native habitats (including the Mediterraneaen Sea from the Red Sea). They then deeply characterized the firefish transposable element (TE) landscape and, by including it in a phylogenetic framework with >40 other fishes, carried out a series of comparative genomic analyses in order to investigate the occurrence of similarities/differences between different genomic features in P. miles.
This paper is very well written and easy to follow, the analyses performed are robust, extensively explained and rich of methodological details. The programs and codes used, both available and devised appositely for this study, are appropriately described along the text and provided in publicly accessible repositories.
The genome assembly statistics are good, with excellent levels of completeness and good contiguity metrics. Overall, this is a rigorous study reporting the genome assembly of an ecologically key species, especially in the light of global warming that will further favor its success as invasive species. Such high-quality genome will help future studies that will rely, for example, on population-based individual resequencing to investigate the genomic consequences of such biological invasions.
That said, I only I have a few minor comments that I hope will help to improve this study:
1) Is anything known about the karyotype of this species? If the karyotype is known, it could be interesting to compare the number of near-chromosomes assembled with the real number of chromosomes to have another estimate of the quality (contiguity in this case) of the assembly.
2) On the same lines, I see that the authors, when describing the pipeline used for the genome assembly with the software Flye, mentioned a “genome size estimation of 900Mb”. Did the author estimate genome size in this study? This can be quickly done with a Kmer-based approach (e.g., with GenomeScope) using the Illumina reads, and might represent a nice addition to this study.
3) The authors carried out a very rigorous analysis to characterize the TE landscape of this fish species, and I very much like the approach they used. It is striking to see that TEs make a large part of the genome (>46%), more than usually seen in fishes with similar genome size. I am sure it is quite difficult/impossible to interpret the functional meaning of such TE expansion in this lineage, but I would like to see something about the expansion history of these TEs. It would be interesting to check whether such expansion was old or recent, and which TE classes expanded in certain times. Kimura substitution levels could be used to calculate and then draw the TE history.
4) Why do not perform a positive selection analysis on the whole gene set? The authors generated all the required input files and the appropriate phylogenetic framework, the same used for the gene contraction/expansion analysis. With some little more work one could investigate positive selection acting on the genes in the firefish lineage, and then making gene function analysis (e.g., GO terms) on the candidate genes found to be under positive selection. This could be discussed in the light of the lineage-specific/unique phenotypes of this species (for example the spines).
5) I would make Fig. 1 and Fig. 2 supplementary material, there is not critical information for the main message of this study here, only technical details in the former, little information than can be included in the text in the latter. Another nice main figure, possibly included in a muti-panel figure with the current Fig. 4, can be the TE history plot (see my comment nr. 3 above).
6) “Table 7. Completeness assessment (%)…”: Is this a BUSCO completeness assessment? I would make it clear in the table legend.
https://doi.org/10.24072/pci.genomics.100241.rev11Kitsoulis and collaborators provide the assembly and annotation of the devil firefish genome at a near-chromosome level,being the first available assembly of the family Scorpaenidae. The importance of this valuable reference genome is highlighted due to the invasive nature of the species, and therefore the need to generate the reference genome that provides the missing resource to study the biology, ecology and phylogeny of this successful invader.
The methodology is adequate for the main goal of the study. The combination of long-read MinION sequencing with more accurate Illumina short reads is a good approach to obtain a high-quality and complete genome assembly, as confirmed by the statistics reported in the article, including the N50,the BUSCO score and the circos plot showing the synteny with the 21 chromosomes of G. aculeatus . The maximum likelihood phylogenetic tree shows highly supported nodes placing the devil firefish within the Perciformes clade, being the first using genomic data. Regarding the genome structural and functional annotation, the implementation of three approaches yielded a very complete set of gene models. The characterization of TEs and gene losses and duplications is also very complete, providing a valuable resource for studying the role of these genomic variations in evolution and the adaptive potential of an invasive species like the devil firefish.
Overall, I consider that the study is very interesting and the methodology implemented is correct and matches the goals of the research. Therefore, as far as I am concerned, I find this article to be scientifically significant and technically accurate. I would just like to propose a few minor changes/recommendations to facilitate the reading.
I would mention the Tables and figures in order. For instance, the first table and figure that are mentioned are table and figure 3. I would reorder the tables to match the appearance in the text.
In order to reduce the number of figures, I would consider moving Figure 3 to supplementary materials and including the L50 in Table 3 (that as I mentioned in the previous comment it should be table 1).
https://doi.org/10.24072/pci.genomics.100241.rev12
