
Recommendation
One important goal of modern genetics is to establish functional associations between genotype and phenotype. Single nucleotide polymorphisms (SNPs) are numerous and widely distributed in the genome and can be obtained from nucleic acid sequencing (1). SNPs allow for the investigation of genetic diversity, which is critical for increasing crop resilience to the challenges posed by global climate change. The associations between SNPs and phenotypes can be captured in genome-wide association studies. SNPs can also be used in combination with machine learning, which is becoming more popular for predicting complex phenotypic traits like yield and biotic and abiotic stress tolerance from genotypic data (2). The availability of many SNP datasets is important in machine learning predictions because this approach requires big data to build a comprehensive model of the association between genotype and phenotype.
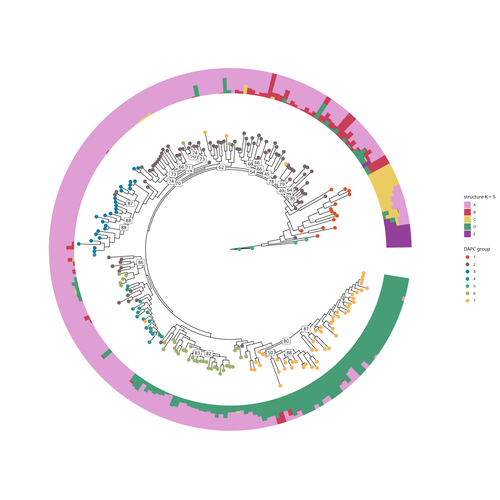
Aubert and colleagues have studied, as part of the PeaMUST project, the genetic diversity of 240 Pisum accessions (3). They sequenced exome-enriched genomic libraries, a technique that enables the identification of high-density, high-quality SNPs at a low cost (4). This technique involves capturing and sequencing only the exonic regions of the genome, which are the protein-coding regions. A total of 2,285,342 SNPs were obtained in this study. The analysis of these SNPs with the annotations of the genome sequence of one of the studied pea accessions (5) identified a number of SNPs that could have an impact on gene activity. Additional analyses revealed 647,220 SNPs that were unique to individual pea accessions, which might contribute to the fitness and diversity of accessions in different habitats. Phylogenetic and clustering analyses demonstrated that the SNPs could distinguish Pisum germplasms based on their agronomic and evolutionary histories. These results point out the power of selected SNPs as markers for identifying Pisum individuals.
Overall, this study found high-quality SNPs that are meaningful in a biological context. This dataset was derived from a large set of germplasm and is thus particularly useful for studying genotype-phenotype associations, as well as the diversity within Pisum species. These SNPs could also be used in breeding programs to develop new pea varieties that are resilient to abiotic and biotic stressors.
References
1. Fallah M, Jean M, Boucher St-Amour VT, O’Donoughue L, Belzile F. The construction of a high-density consensus genetic map for soybean based on SNP markers derived from genotyping-by-sequencing. Genome. 2022 Aug;65(8):413–25.
https://doi.org/10.1139/gen-2021-005
2. Gill M, Anderson R, Hu H, Bennamoun M, Petereit J, Valliyodan B, et al. Machine learning models outperform deep learning models, provide interpretation and facilitate feature selection for soybean trait prediction. BMC Plant Biology. 2022 Apr 8;22(1):180.
https://doi.org/10.1186/s12870-022-03559-z
3. Aubert G, Kreplak J, Leveugle M, Duborjal H, Klein A, Boucherot K, et al. SNP discovery by exome capture and resequencing in a pea genetic resource collection., biorxiv, ver. 4, peer-reviewed and recommended by Peer Community in Genomics.
https://doi.org/10.1101/2022.08.03.502586
4. Warr A, Robert C, Hume D, Archibald A, Deeb N, Watson M. Exome sequencing: current and future perspectives. G3 Genes|Genomes|Genetics. 2015 Aug 1;5(8):1543–50.
https://doi.org/10.1534/g3.115.018564
5. Kreplak J, Madoui MA, Cápal P, Novák P, Labadie K, Aubert G, et al. A reference genome for pea provides insight into legume genome evolution. Nat Genet. 2019 Sep;51(9):1411–22.
DOI or URL of the preprint: https://doi.org/10.1101/2022.08.03.502586
Version of the preprint: 3
Reviewers are highly satisfied with your response to their suggestions. The quality of the revised document was sufficient for publication. However, one of the reviewers had a few additional suggestions, which are very valuable. Please address these two final suggestions.
The authors have addressed all my suggestions, and I now recommend to accept the revised manuscript.
https://doi.org/10.24072/pci.genomics.100237.rev21 , 05 Jun 2023
, 05 Jun 2023During the initial round of revisions for Aubert's research article entitled "SNP discovery by exome capture and resequencing in a pea genetic resource collection," I expressed a significant concern regarding the phylogenetic analyses, accompanied by several minor concerns. I am pleased to report that all of these concerns have been effectively addressed. The authors employed a coalescence-based phylogenetic inference in conjunction with the standard substitution model, leading to the observation of variations in the clades, which were attributed to adixture leading to incomplete lineage sorting (ILS).
Nevertheless, I would like to propose two final suggestions.
1. Firstly, I would recommend the authors to incorporate clade support measures within their tree representations. While acknowledging that the authors are preparing a subsequent paper dedicated to comprehensive phylogenetic analyses, it is important to acknowledge that once a phylogenetic tree is published, it often becomes the foundation for subsequent analyses, even when certain clades may exhibit lower resolution. In light of the present study's indication of disparate outcomes between the two employed methods, likely due to ILS, providing measures of clade support derived from these methods becomes important. Such information would empower potential users of the analyses to assess the robustness of these discrepancies and make more informed decisions.
2. Additionally, I suggest highlighting the specification of the two methods used, namely the standard substitution model and the coalescence-based approach, in line 143.
Overall, I express my satisfaction with the revisions implemented in this manuscript.
Rui Borges (Institute of Population Genetics, Vetmeduni Vienna)
https://doi.org/10.24072/pci.genomics.100237.rev22
DOI or URL of the preprint: https://doi.org/10.1101/2022.08.03.502586
Version of the preprint: 2
The preprint entitled "SNP discovery by exome capture and resequencing in a pea genetic resource collection ", which you recently submitted to the Peer Community in Genomics (PCI Genomics), has now been reviewed. The decision on this manuscript was: Revision. Therefore, I invite you to respond to the reviewers' comments shown below and revise your manuscript accordingly.
, 31 Dec 2022The paper by Aubert, "SNP discovery by exome capture and resequencing in a pea genetic resource collection," aims to evaluate the genetic diversity of a collection of 240 pea (Pisum sativum L.) accessions. This paper reports on the large number of SNPs (approximately 2.3 million) obtained through whole-exome sequencing. The methods described in the paper appear to be sufficiently detailed. Additionally, I agree with the authors' conclusions and recognize the potential value of this dataset for future genome-wide association studies and breeding programs in pea.
My main concern is with the phylogenetic analysis. The authors carried out their analysis using SNPs while ignoring constant sites (lines 92-94). This could potentially impact the estimated distances based on the GTR model. It is not clear to me if this will significantly affect the tree topology (although, I suspect it will not), but it could alter the branch lengths. I recommend that the authors take steps to correct for ascertainment bias in their analyses. Furthermore, the authors chose to present the tree as a cladogram, which does not allow the reader to appreciate the branch lengths. This information could be useful in understanding the differences found between the clustering and structure methods (lines 152-53). For example, the authors should consider whether long-branch attraction could be a factor.
My primary concern, however, is more fundamental. Given that the authors are working with genetic positions that are not fully sorted, can they accurately estimate a phylogenetic tree based on the GTR distance? I am uncertain how to interpret the estimated clades and distances in this context. Therefore, I believe it would be more appropriate to estimate a coalescent tree instead, as it provides a more appropriate description of the dataset the authors have on hand. This is of fundamental importance because I believe that some of the clades estimated in this work are likely to be used in further studies.
There are also a few minor aspects I would like to address:
Rui Borges (Institute of Population Genetics, Vetmeduni Vienna)
https://doi.org/10.24072/pci.genomics.100237.rev11In the manuscript “SNP discovery by exome capture and resequencing in a pea genetic resource collection”, the authors performed exome sequencing to genotype 240 accessions of pea and identified a dataset of SNPs to be used for genetic diversity analysis. For this, the authors selected a large number of samples, including cultivars, landraces, and wild types with diverse geographical origins that follow the standard approach of population genetic analysis. This study is interesting and has valuable information, but some details still need to be improved. The following are some questions and suggestions for modification:
