
LERAT Emmanuelle
- BIométrie et Biologie Evolutive, CNRS - Université Lyon 1, Villeurbanne, France
- Bioinformatics, Evolutionary genomics, Viruses and transposable elements
- recommender
Recommendations: 2
Reviews: 0
Recommendations: 2
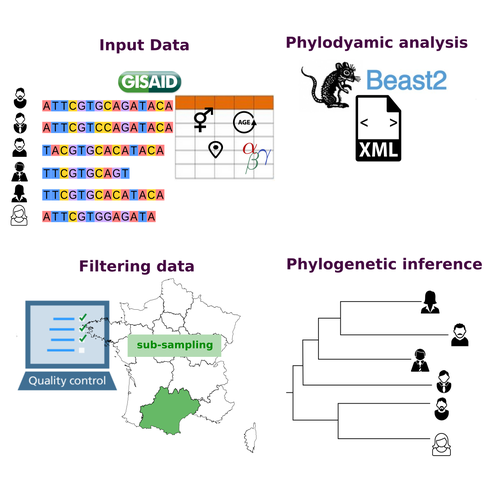
COVFlow: phylodynamics analyses of viruses from selected SARS-CoV-2 genome sequences
A pipeline to select SARS-CoV-2 sequences for reliable phylodynamic analyses
Recommended by Emmanuelle Lerat based on reviews by Gabriel Wallau and Bastien BoussauPhylodynamic approaches enable viral genetic variation to be tracked over time, providing insight into pathogen phylogenetic relationships and epidemiological dynamics. These are important methods for monitoring viral spread, and identifying important parameters such as transmission rate, geographic origin and duration of infection [1]. This knowledge makes it possible to adjust public health measures in real-time and was important in the case of the COVID-19 pandemic [2]. However, these approaches can be complicated to use when combining a very large number of sequences. This was particularly true during the COVID-19 pandemic, when sequencing data representing millions of entire viral genomes was generated, with associated metadata enabling their precise identification.
Danesh et al. [3] present a bioinformatics pipeline, CovFlow, for selecting relevant sequences according to user-defined criteria to produce files that can be used directly for phylodynamic analyses. The selection of sequences first involves a quality filter on the size of the sequences and the absence of unresolved bases before being able to make choices based on the associated metadata. Once the sequences are selected, they are aligned and a time-scaled phylogenetic tree is inferred. An output file in a format directly usable by BEAST 2 [4] is finally generated.
To illustrate the use of the pipeline, Danesh et al. [3] present an analysis of the Delta variant in two regions of France. They observed a delay in the start of the epidemic depending on the region. In addition, they identified genetic variation linked to the start of the school year and the extension of vaccination, as well as the arrival of a new variant. This tool will be of major interest to researchers analysing SARS-CoV-2 sequencing data, and a number of future developments are planned by the authors.
References
[1] Baele G, Dellicour S, Suchard MA, Lemey P, Vrancken B. 2018. Recent advances in computational phylodynamics. Curr Opin Virol. 31:24-32. https://doi.org/10.1016/j.coviro.2018.08.009
[2] Attwood SW, Hill SC, Aanensen DM, Connor TR, Pybus OG. 2022. Phylogenetic and phylodynamic approaches to understanding and combating the early SARS-CoV-2 pandemic. Nat Rev Genet. 23:547-562. https://doi.org/10.1038/s41576-022-00483-8
[3] Danesh G, Boennec C, Verdurme L, Roussel M, Trombert-Paolantoni S, Visseaux B, Haim-Boukobza S, Alizon S. 2023. COVFlow: phylodynamics analyses of viruses from selected SARS-CoV-2 genome sequences. bioRxiv, ver. 7 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2022.06.17.496544
[4] Bouckaert R, Heled J, Kühnert D, Vaughan T, Wu C-H et al. 2014. BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput Biol 10: e1003537. https://doi.org/10.1371/journal.pcbi.1003537
A pipeline to detect the relationship between transposable elements and adjacent genes in host genomes
A new tool to cross and analyze TE and gene annotations
Recommended by Emmanuelle Lerat based on reviews by 2 anonymous reviewersTransposable elements (TEs) are important components of genomes. Indeed, they are now recognized as having a major role in gene and genome evolution (Biémont 2010). In particular, several examples have shown that the presence of TEs near genes may influence their functioning, either by recruiting particular epigenetic modifications (Guio et al. 2018) or by directly providing new regulatory sequences allowing new expression patterns (Chung et al. 2007; Sundaram et al. 2014). Therefore, the study of the interaction between TEs and their host genome requires tools to easily cross-annotate both types of entities. In particular, one needs to be able to identify all TEs located in the close vicinity of genes or inside them. Such task may not always be obvious for many biologists, as it requires informatics knowledge to develop their own script codes.
In their work, Meguerdichian et al. (2021) propose a command-line pipeline that takes as input the annotations of both genes and TEs for a given genome, then detects and reports the positional relationships between each TE insertion and their closest genes. The results are processed into an R script to provide tables displaying some statistics and graphs to visualize these relationships.
This tool has the potential to be very useful for performing preliminary analyses before studying the impact of TEs on gene functioning, especially for biologists. Indeed, it makes it possible to identify genes close to TE insertions. These identified genes could then be specifically considered in order to study in more detail the link between the presence of TEs and their functioning. For example, the identification of TEs close to genes may allow to determine their potential role on gene expression.
References
Biémont C (2010). A brief history of the status of transposable elements: from junk DNA to major players in evolution. Genetics, 186, 1085–1093. https://doi.org/10.1534/genetics.110.124180
Chung H, Bogwitz MR, McCart C, Andrianopoulos A, ffrench-Constant RH, Batterham P, Daborn PJ (2007). Cis-regulatory elements in the Accord retrotransposon result in tissue-specific expression of the Drosophila melanogaster insecticide resistance gene Cyp6g1. Genetics, 175, 1071–1077. https://doi.org/10.1534/genetics.106.066597
Guio L, Vieira C, González J (2018). Stress affects the epigenetic marks added by natural transposable element insertions in Drosophila melanogaster. Scientific Reports, 8, 12197. https://doi.org/10.1038/s41598-018-30491-w
Meguerditchian C, Ergun A, Decroocq V, Lefebvre M, Bui Q-T (2021). A pipeline to detect the relationship between transposable elements and adjacent genes in host genomes. bioRxiv, 2021.02.25.432867, ver. 4 peer-reviewed and recommended by Peer Community In Genomics. https://doi.org/10.1101/2021.02.25.432867
Sundaram V, Cheng Y, Ma Z, Li D, Xing X, Edge P, Snyder MP, Wang T (2014). Widespread contribution of transposable elements to the innovation of gene regulatory networks. Genome Research, 24, 1963–1976. https://doi.org/10.1101/gr.168872.113