
Recommendation
Contamination, the presence of foreign DNA sequences in a sample of interest, is currently a major problem in genomics. Because contamination is often unavoidable at the experimental stage, it is increasingly recognized that the processing of high-throughput sequencing data must include a decontamination step. This is usually performed after the many sequence reads have been assembled into a relatively small number of contigs. Dubious contigs are then discarded based on their composition (e.g. GC-content) or because they are highly similar to a known piece of DNA from a foreign species.
Here [1], Mathieu Gautier explores a novel strategy consisting in decontaminating reads, not contigs. Why is this promising? Assembly programs and algorithms are complex, and it is not easy to predict, or monitor, how they handle contaminant reads. Ideally, contaminant reads will be assembled into obvious contaminant contigs. However, there might be more complex situations, such as chimeric contigs with alternating genuine and contaminant segments. Decontaminating at the read level, if possible, should eliminate such unfavorable situations where sequence information from contaminant and target samples are intimately intertwined by an assembler.
To achieve this aim, Gautier proposes to use methods initially designed for the analysis of metagenomic data. This is pertinent since the decontamination process involves considering a sample as a mixture of different sources of DNA. The programs used here, CLARK and CLARK-L, are based on so-called k-mer analysis, meaning that the similarity between a read to annotate and a reference sequence is measured by how many sub-sequences (of length 31 base pairs for CLARK and 27 base pairs for CLARK-L) they share. This is notoriously more efficient than traditional sequence alignment algorithms when it comes to comparing a very large number of (most often unrelated) sequences. This is, therefore, a reference-based approach, in which the reads from a sample are assigned to previously sequenced genomes based on k-mer content.
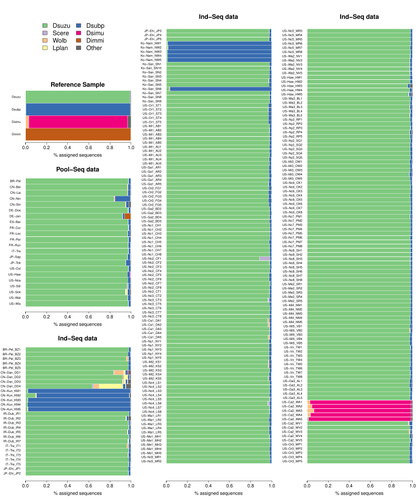
This original approach is here specifically applied to the case of Drosophila suzukii, an invasive pest damaging fruit production in Europe and America. Fortunately, Drosophila is a genus of insects with abundant genomic resources, including high-quality reference genomes in dozens of species. Having calibrated and validated his pipeline using data sets of known origins, Gautier quantifies in each of 258 presumed D. suzukii samples the proportion of reads that likely belong to other species of fruit flies, or to fruit fly-associated microbes. This proportion is close to one in 16 samples, which clearly correspond to mis-labelled individuals. It is non-negligible in another ~10 samples, which really correspond to D. suzukii individuals. Most of these reads of unexpected origin are contaminants and should be filtered out. Interestingly, one D. suzukii sample contains a substantial proportion of reads from the closely related D. subpulchera, which might instead reflect a recent episode of gene flow between these two species. The approach, therefore, not only serves as a crucial technical step, but also has the potential to reveal biological processes.
Gautier's thorough, well-documented work will clearly benefit the ongoing and future research on D. suzuki, and Drosophila genomics in general. The author and reviewers rightfully note that, like any reference-based approach, this method is heavily dependent on the availability and quality of reference genomes - Drosophila being a favorable case. Building the reference database is a key step, and the interpretation of the output can only be made in the light of its content and gaps, as illustrated by Gautier's careful and detailed discussion of his numerous results.
This pioneering study is a striking demonstration of the potential of metagenomic methods for the decontamination of high-throughput sequence data at the read level. The pipeline requires remarkably few computing resources, ensuring low carbon emission. I am looking forward to seeing it applied to a wide range of taxa and samples.
Reference
[1] Gautier Mathieu. Efficient k-mer based curation of raw sequence data: application in Drosophila suzukii. bioRxiv, 2023.04.18.537389, ver. 2, peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2023.04.18.537389
DOI or URL of the preprint: https://doi.org/10.1101/2023.04.18.537389
Version of the preprint: 1
This study introduces a novel approach for assessing and treating the problems of contamination and mislabeling in high-throughput genomic data. The idea is to recycle methods developed for species identification based on metagenomic data. The manuscript was reviewed by two colleagues, both of which are very positive - and so am I. A number of relevant suggestions were made, which I think should help improve the manuscript.
I have an aditional comment, also briefly mentioned by one reviewer. Besides contamination, there might be biological reasons why a given sample contains sequence reads assigned to a different species, namely hybridization and gene flow. How is the newly introduced method expected to behave when reproductive isolation between the analyzed species is incomplete? In particular, is there a risk that the method partially erases the signal of gene flow, if actually present? I think these questions could deserve a specific discussion as gene flow is quite common in nature and the focus of many population genomic studies.
I would be happy to consider a revised version of the manuscript for possible recommendation.
This article describes a procedure used to control publicly available sequence data of Drosophila Suzukii for mislabeling and contaminations. The procedure relies on the construction of discriminatory k-mers dictionaries to compare with k-mers present in each dataset. It was performed using the software CLARK, which was created for the taxonomic classification of metagenomic sequences.
The procedure efficiently identified 16 mislabeled samples among the 236 individual D. suzukii sequence data and 2 contaminated samples among 22 pool-seq sequence data.
I found this approach really interesting and well presented in the manuscript.
The author 1) advocates for the routine inclusion of such k-mer based quality check in data quality assessment practices. 2) presents a curated dataset of D. suzukii public sequences, useful for further population genomics studies.
I may have a question regarding the idea that such check should be included in standard quality assessment. In this analysis, the author relied on extensive and curated assemblies genomic data (« high quality assemblies for several dozen of drosophilid genomes »). Here, these numerous genomes also allow to evaluate the "global" efficiency of the approach, but I wonder to what extend such approach could be easily generalized for any species. What would be the author guidelines to perform such check for any genomic dataset ? To say it differently, what would be the minimal external data (in terms of both quality of assembly and taxonomic coverage) required to construct a meaningful dictionary ?
L47-51 the repetition of « the resulting combined datasets » might be avoided.
L237. I think « 305 » should be « 301 », to match the sum listed in the paragraph (43+236 +22), which is also coherent with the number of lines in table S2 and S3 and to the value L331.
Sorry if I missed the correct sens of the number.
Fig 2B . Are the colors corresponding to target and other (light and dark blue) reversed? I expected the more dispersed and almost bimodal distribution (dark blue), with higher percentage of sequences with no match to correspond to the « other species ».
L314-316 Does this option -s 2 have a strong impact on computation time and fraction of sequences with no matching k-mers? ?
l410 « may thus [be] display »
I was able to retrieve the databases, cleaned assemblies and scripts from the Data INRAE repository but I did not attempted to run clark myself. However, they look well formatted and organized.
In “run_fastp_clarkl_clark_and_summarize_results.sh”: l20: “cleanning seqeunce” → “cleaning sequences”
https://doi.org/10.24072/pci.genomics.100244.rev11In this empirical study on Drosophila whole genome samples, Gautier evaluates the use of the metagenomic classifier CLARK to analyse the contamination structure of short-read datasets by closely related species of the advertised organism and its microbial commensals. The author shows that this approach is both accurate and computationally efficient and, as a byproduct, releases a curated set of >60 population samples of D. suzuki that should be useful in future population genetic studies.
Generally speaking, I enjoyed reviewing this manuscript. The study is well-designed, the text is clear and pleasant to read and the figures are easy to understand. Moreover, the work is extremely well-documented, with most of the study details provided in Supplementary Tables, while data and scripts are made available in a public repository (please note that I did not download the latter to check the actual content). Consequently, my comments are minor and aimed at further clarifying the text when needed. However, I noticed a number of small errors in the reporting of the results. As some of them are quite confusing, I insist that they should be addressed in the revision of the manuscript.
* Scientific questions
- lines 173-175: I don't understand if the 101 assemblies of the paper (which are taxonomically diverse) are part of the 129 assemblies on the NCBI portal and, if not, why the former were not preferred to the latter? Was there some global quality assessment of all available assemblies (in the NCBI and elsewhere) prior to taking these decisions?
- lines 197-198 ("widespread lateral gene transfer from Wolbachia"): this raises the issue of whether such transfers should be considered as contamination in this species... and in other species! On a side note, had the species datasets completely devoid of Wolbachia sequences been aggressively curated before public release?
- lines 209 ("after filtering out contaminating sequences"): if I understand correctly, Kraken2 was used on whole contigs, not pseudo-reads spliced out of contigs. Then does "filtering out" mean removing these whole contigs (i.e., up to 1.4 Mb in one case)? Was it not possible to preserve more information by only masking the foreign regions of large contigs (assuming they might be chimeric)?
- lines 213-216: it is mentioned briefly in the Discussion (lines 766-769 and 793-795), but I wonder if "pangenomes" (rather than single strains) would have provided more sensitivity for pathogen and commensal screening. This is an important issue from a practical point of view.
- lines 243-244 ("including data on 12 of the 29 target species"): is it on purpose that 17 of the target species are not tested by the samples?
- in Table 1: I know that it is suggested in CLARK paper, but I wonder if the representation of some species by multiple assemblies is really harmless in terms of assignment statistics. Similarly, are we sure that the results are not biased in some way when some species are more distant and thus would provide a lot more specific k-mers than groups of highly related species? I did not find a discussion of this issue in CLARK paper, but for the present purposes, knowing the answer would be important. If so, it might be introduced at lines 298-300. Also, a related bit of discussion appears at lines 665-677.
- lines 478-494: for the 16 species not represented in the target dictionaries but still assigned to a single target species, 5 are assigned to D. bipectinata (and none to D. ananassae) and 2 to D. obscura (and none to D. subobscura). Is there a phylogenetic reason for this?
* Clarity issues
- lines 16-31 in the Abstract are a copy-paste from the end of the Introduction (lines 137-152); maybe rephrase some sentences?
- lines 57-58 ("the characteristics mentioned above have mostly remained"): I don't understand the idea here; please rephrase.
- lines 87-88 ("but they are not well suited for the analysis of large amounts of samples"): please add a hint about why it is so.
- line 91 ("the genomes of the putative contaminant species"): this is a bit restrictive (only negative filtering), especially considering that the current study use both positive and negative filtering; please add a bit of nuance. BTW, positive filtering is discussed at lines 700-704.
- lines 159-160: please explain the logic behind the phylogenetic breadth of the reference sampling to help others (e.g., why also the subgenus Drosophila).
- lines 161-163 ("for subgroups or groups represented by multiple assemblies, only one species was selected"): ambiguous phrasing: multiple assemblies of the same species or multiple assemblies of different species? In my view, one assembly does not always equate one species.
- line 186 ("including Wolbachia endosymbionts"): ambiguous wording; is it an exception or a precision?
- lines 230-232 ("Building the k–mer dictionary took 2h46min"): such timings are quite useless without some idea of the CPU architecture; please specify it.
- line 307 (and around): in CLARK paper, the confidence score is only computed based on the two top-matching sequences, not all; please check.
- lines 349-350 ("sequence length was representative of typical short read datasets"); please state that datasets here include a variable mixture of merged and unmerged reads (if I understand correctly).
- line 379 ("averaging 24.5%"): why to report a mean here and everywhere else median values? Is there a specific reason?
- Tables S4/S5 (and lines 414-415): "assignable (and assigned) sequences" should be better defined (see also my comment below for line 325). "% assigned sequences (with at least one matching kmer)" in head of Col E is confusing because either a) it should complement Col D "% seq with no matching kmer" [since a sequence either has zero or at least one matching k-mer (= assignable?)] or b) Col E actually reports the fraction of assignable sequences that are assigned (at >=5/6 and >=0.95 thresholds?). Please clarify.
- in legend of Figure 2 ("corresponding target dictionary"): why "corresponding" here? There is only one global dictionary per method, correct?
- lines 503-505 ("capture less than 30% of the assigned sequences"): the text does not exactly match what is shown in Figure S4 (rather 40% for Doshi, Dprui and Dbock while Dcard is not cited). Why such a discrepancy with Figure 3?
- lines 527-529: if I count correctly, 5 Ind-Seq samples are not mentioned in this part (236-215-16 = 5). Four of them are cited when discussing Wolbachia contamination, but not the last one: US-Nc2_CF1. Anything to say about it?
- lines 708-710 (about filtering based on k-mers): I agree with the assertion, but it seems ironic that target contigs were filtered with Kraken2 in the present study. It should be explicitly reminded here to avoid the feeling.
- lines 732-737 (about contaminated Pool-Seq samples): was this issue known prior to the current study? If not, this would be useful to state it.
- legend of Figure S2: why "Total assignment time"? I guess it includes sample loading time, but this is not mentioned in the main text. Is it what this means?
* Mild suggestions
- line 142 (and elsewhere): were assigned => were re-assigned [to emphasize the original assignment error?]
- lines 184,189-190: choose between "contaminating" and "contaminated"? In the present case, they are used interchangeably and this might be confusing.
- Figure 1: why two species names in bold? Besides, for consistency with, e.g., willistoni, I would add the subgroup guinaria and virilis in the figure (especially because "subgroup virilis" is used in the text).
- lines 494-495: these samples => these 16 samples [for clarity and maybe it would be useful to color them differently in Figure S4]
- lines 499-500: the most represented species => the most closely related represented species [also check y axis in Figure S4].
- line 539: 1. 71% => 1.71%
- Figure S1B (y axis): %% overlapping => % overlapping
* Reporting errors
- line 6: 32 => 22
- line 114: n=8 => n=6
- in the Excel file, Tables S2 and S3 are reversed
- line 250: n=3 => n=4 [and "missing Illumina HiSeq X Ten (PE150) (n=1)"]
- line 261 ("all sequenced on an Illumina HiSeq4000 in PE150 mode") => except 30 samples sequenced in PE100
- Tables S2/S3: column headers for timing values are incorrect, which makes the section about run times extremely confusing; please check and fix! Moreover, the head of the column for overlapping values has the word "Non" in it, which (wrongly) suggests that these numbers are "non-overlapping reads" (see also line 281 in main text).
- line 325 (and elsewhere): I am not sure that it is an error, but to me, >1 and >5 mean "at least two" and "at least 6", respectively. Is it what is meant here? The issue is important because the section about the proportion of assigned sequences is difficult to understand with this doubt in mind (see comment above).
- Figure 2B: I am pretty sure that there is an error in the order of the first two violin plots. Target sp and Other sp are probably reversed because, as such, they neither match the text (lines 389-398) nor Figure 2A. Color key is right though. Please check and fix!
