
Latest recommendations

| Id | Title▼ | Authors | Abstract | Picture | Thematic fields | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
27 Apr 2021

Uncovering transposable element variants and their potential adaptive impact in urban populations of the malaria vector Anopheles coluzziiAnopheles coluzzii, a new system to study how transposable elements may foster adaptation to urban environmentsRecommended by Anne Roulin based on reviews by Yann Bourgeois and 1 anonymous reviewerTransposable elements (TEs) are mobile DNA sequences that can increase their copy number and move from one location to another within the genome [1]. Because of their transposition dynamics, TEs constitute a significant fraction of eukaryotic genomes. TEs are also known to play an important functional role and a wealth of studies has now reported how TEs may influence single host traits [e.g. 2–4]. Given that TEs are more likely than classical point mutations to cause extreme changes in gene expression and phenotypes, they might therefore be especially prone to produce the raw diversity necessary for individuals to respond to challenging environments [5,6] such as the ones found in urban area.
| Uncovering transposable element variants and their potential adaptive impact in urban populations of the malaria vector Anopheles coluzzii | Carlos Vargas-Chavez, Neil Michel Longo Pendy, Sandrine E. Nsango, Laura Aguilera, Diego Ayala, and Josefa González | <p style="text-align: justify;">Background</p> <p style="text-align: justify;">Anopheles coluzzii is one of the primary vectors of human malaria in sub-Saharan Africa. Recently, it has colonized the main cities of Central Africa threatening vecto... | | Evolutionary genomics | Anne Roulin | 2020-12-02 14:58:47 | ||
19 Jul 2021
TransPi - a comprehensive TRanscriptome ANalysiS PIpeline for de novo transcriptome assemblyTransPI: A balancing act between transcriptome assemblersRecommended by Oleg Simakov based on reviews by Gustavo Sanchez and Juan Daniel Montenegro CabreraEver since the introduction of the first widely usable assemblers for transcriptomic reads (Huang and Madan 1999; Schulz et al. 2012; Simpson et al. 2009; Trapnell et al. 2010, and many more), it has been a technical challenge to compare different methods and to choose the “right” or “best” assembly. It took years until the first widely accepted set of benchmarks beyond raw statistical evaluation became available (e.g., Parra, Bradnam, and Korf 2007; Simão et al. 2015). However, an approach to find the right balance between the number of transcripts or isoforms vs. evolutionary completeness measures has been lacking. This has been particularly pronounced in the field of non-model organisms (i.e., wild species that lack a genomic reference). Often, studies in this area employed only one set of assembly tools (the most often used to this day being Trinity, Haas et al. 2013; Grabherr et al. 2011). While it was relatively straightforward to obtain an initial assembly, its validation, annotation, as well its application to the particular purpose that the study was designed for (phylogenetics, differential gene expression, etc) lacked a clear workflow. This led to many studies using a custom set of tools with ensuing various degrees of reproducibility. TransPi (Rivera-Vicéns et al. 2021) fills this gap by first employing a meta approach using several available transcriptome assemblers and algorithms to produce a combined and reduced transcriptome assembly, then validating and annotating the resulting transcriptome. Notably, TransPI performs an extensive analysis/detection of chimeric transcripts, the results of which show that this new tool often produces fewer misassemblies compared to Trinity. TransPI not only generates a final report that includes the most important plots (in clickable/zoomable format) but also stores all relevant intermediate files, allowing advanced users to take a deeper look and/or experiment with different settings. As running TransPi is largely automated (including its installation via several popular package managers), it is very user-friendly and is likely to become the new "gold standard" for transcriptome analyses, especially of non-model organisms. References Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature Biotechnology, 29, 644–652. https://doi.org/10.1038/nbt.1883 Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, Couger MB, Eccles D, Li B, Lieber M, MacManes MD, Ott M, Orvis J, Pochet N, Strozzi F, Weeks N, Westerman R, William T, Dewey CN, Henschel R, LeDuc RD, Friedman N, Regev A (2013) De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nature Protocols, 8, 1494–1512. https://doi.org/10.1038/nprot.2013.084 Huang X, Madan A (1999) CAP3: A DNA Sequence Assembly Program. Genome Research, 9, 868–877. https://doi.org/10.1101/gr.9.9.868 Parra G, Bradnam K, Korf I (2007) CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics, 23, 1061–1067. https://doi.org/10.1093/bioinformatics/btm071 Rivera-Vicéns RE, Garcia-Escudero CA, Conci N, Eitel M, Wörheide G (2021) TransPi – a comprehensive TRanscriptome ANalysiS PIpeline for de novo transcriptome assembly. bioRxiv, 2021.02.18.431773, ver. 3 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2021.02.18.431773 Schulz MH, Zerbino DR, Vingron M, Birney E (2012) Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics, 28, 1086–1092. https://doi.org/10.1093/bioinformatics/bts094 Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM (2015) BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics, 31, 3210–3212. https://doi.org/10.1093/bioinformatics/btv351 Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJM, Birol İ (2009) ABySS: A parallel assembler for short read sequence data. Genome Research, 19, 1117–1123. https://doi.org/10.1101/gr.089532.108 Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L (2010) Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature Biotechnology, 28, 511–515. https://doi.org/10.1038/nbt.1621 | TransPi - a comprehensive TRanscriptome ANalysiS PIpeline for de novo transcriptome assembly | Ramon E Rivera-Vicens, Catalina Garcia-Escudero, Nicola Conci, Michael Eitel, Gert Wörheide | <p style="text-align: justify;">The use of RNA-Seq data and the generation of de novo transcriptome assemblies have been pivotal for studies in ecology and evolution. This is distinctly true for non-model organisms, where no genome information is ... | | Bioinformatics, Evolutionary genomics | Oleg Simakov | 2021-02-18 20:56:08 | ||
18 Feb 2021
Traces of transposable element in genome dark matter co-opted by flowering gene regulation networksUsing small fragments to discover old TE remnants: the Duster approach empowers the TE detectionRecommended by Francois Sabot based on reviews by Josep Casacuberta and 1 anonymous reviewer based on reviews by Josep Casacuberta and 1 anonymous reviewer
Transposable elements are the raw material of the dark matter of the genome, the foundation of the next generation of genes and regulation networks". This sentence could be the essence of the paper of Baud et al. (2021). Transposable elements (TEs) are endogenous mobile genetic elements found in almost all genomes, which were discovered in 1948 by Barbara McClintock (awarded in 1983 the only unshared Medicine Nobel Prize so far). TEs are present everywhere, from a single isolated copy for some elements to more than millions for others, such as Alu. They are founders of major gene lineages (HET-A, TART and telomerases, RAG1/RAG2 proteins from mammals immune system; Diwash et al, 2017), and even of retroviruses (Xiong & Eickbush, 1988). However, most TEs appear as selfish elements that replicate, land in a new genomic region, then start to decay and finally disappear in the midst of the genome, turning into genomic ‘dark matter’ (Vitte et al, 2007). The mutations (single point, deletion, recombination, and so on) that occur during this slow death erase some of their most notable features and signature sequences, rendering them completely unrecognizable after a few million years. Numerous TE detection tools have tried to optimize their detection (Goerner-Potvin & Bourque, 2018), but further improvement is definitely challenging. This is what Baud et al. (2021) accomplished in their paper. They used a simple, elegant and efficient k-mer based approach to find small signatures that, when accumulated, allow identifying very old TEs. Using this method, called Duster, they improved the amount of annotated TEs in the model plant Arabidopsis thaliana by 20%, pushing the part of this genome occupied by TEs up from 40 to almost 50%. They further observed that these very old Duster-specific TEs (i.e., TEs that are only detected by Duster) are, among other properties, close to genes (much more than recent TEs), not targeted by small RNA pathways, and highly associated with conserved regions across the rosid family. In addition, they are highly associated with flowering or stress response genes, and may be involved through exaptation in the evolution of responses to environmental changes. TEs are not just selfish elements: more and more studies have shown their key role in the evolution of their hosts, and tools such as Duster will help us better understand their impact. References Baud, A., Wan, M., Nouaud, D., Francillonne, N., Anxolabéhère, D. and Quesneville, H. (2021). Traces of transposable elements in genome dark matter co-opted by flowering gene regulation networks. bioRxiv, 547877, ver. 5 peer-reviewed and recommended by PCI Genomics.doi: https://doi.org/10.1101/547877 | Traces of transposable element in genome dark matter co-opted by flowering gene regulation networks | Agnes Baud, Mariene Wan, Danielle Nouaud, Nicolas Francillonne, Dominique Anxolabehere, Hadi Quesneville | <p>Transposable elements (TEs) are mobile, repetitive DNA sequences that make the largest contribution to genome bulk. They thus contribute to the so-called 'dark matter of the genome', the part of the genome in which nothing is immediately recogn... | | Bioinformatics, Evolutionary genomics, Functional genomics, Plants, Structural genomics, Viruses and transposable elements | Francois Sabot | Anonymous, Josep Casacuberta | 2020-04-07 17:12:12 | |
16 Dec 2022
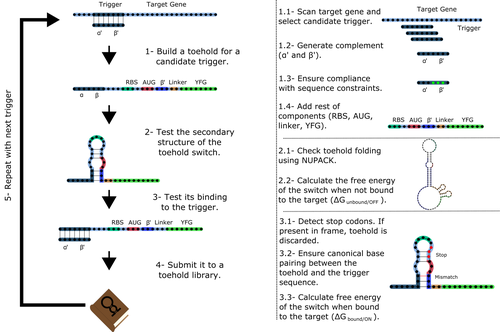
Toeholder: a Software for Automated Design and In Silico Validation of Toehold RiboswitchesA novel approach for engineering biological systems by interfacing computer science with synthetic biologyRecommended by Sahar Melamed based on reviews by Wim Wranken and 1 anonymous reviewerBiological systems depend on finely tuned interactions of their components. Thus, regulating these components is critical for the system's functionality. In prokaryotic cells, riboswitches are regulatory elements controlling transcription or translation. Riboswitches are RNA molecules that are usually located in the 5′-untranslated region of protein-coding genes. They generate secondary structures leading to the regulation of the expression of the downstream protein-coding gene (Kavita and Breaker, 2022). Riboswitches are very versatile and can bind a wide range of small molecules; in many cases, these are metabolic byproducts from the gene’s enzymatic or signaling pathway. Their versatility and abundance in many species make them attractive for synthetic biological circuits. One class that has been drawing the attention of synthetic biologists is toehold switches (Ekdahl et al., 2022; Green et al., 2014). These are single-stranded RNA molecules harboring the necessary elements for translation initiation of the downstream gene: a ribosome-binding site and a start codon. Conformation change of toehold switches is triggered by an RNA molecule, which enables translation. To exploit the most out of toehold switches, automation of their design would be highly advantageous. Cisneros and colleagues (Cisneros et al., 2022) developed a tool, “Toeholder”, that automates the design of toehold switches and performs in silico tests to select switch candidates for a target gene. Toeholder is an open-source tool that provides a comprehensive and automated workflow for the design of toehold switches. While web tools have been developed for designing toehold switches (To et al., 2018), Toeholder represents an intriguing approach to engineering biological systems by coupling synthetic biology with computational biology. Using molecular dynamics simulations, it identified the positions in the toehold switch where hydrogen bonds fluctuate the most. Identifying these regions holds great potential for modifications when refining the design of the riboswitches. To be effective, toehold switches should provide a strong ON signal and a weak OFF signal in the presence or the absence of a target, respectively. Toeholder nicely ranks the candidate toehold switches based on experimental evidence that correlates with toehold performance (based on good ON/OFF ratios). Riboswitches are highly appealing for a broad range of applications, including pharmaceutical and medical purposes (Blount and Breaker, 2006; Giarimoglou et al., 2022; Tickner and Farzan, 2021), thanks to their adaptability and inexpensiveness. The Toeholder tool developed by Cisneros and colleagues is expected to promote the implementation of toehold switches into these various applications. References Blount KF, Breaker RR (2006) Riboswitches as antibacterial drug targets. Nature Biotechnology, 24, 1558–1564. https://doi.org/10.1038/nbt1268 Cisneros AF, Rouleau FD, Bautista C, Lemieux P, Dumont-Leblond N, ULaval 2019 T iGEM (2022) Toeholder: a Software for Automated Design and In Silico Validation of Toehold Riboswitches. bioRxiv, 2021.11.09.467922, ver. 3 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2021.11.09.467922 Ekdahl AM, Rojano-Nisimura AM, Contreras LM (2022) Engineering Toehold-Mediated Switches for Native RNA Detection and Regulation in Bacteria. Journal of Molecular Biology, 434, 167689. https://doi.org/10.1016/j.jmb.2022.167689 Giarimoglou N, Kouvela A, Maniatis A, Papakyriakou A, Zhang J, Stamatopoulou V, Stathopoulos C (2022) A Riboswitch-Driven Era of New Antibacterials. Antibiotics, 11, 1243. https://doi.org/10.3390/antibiotics11091243 Green AA, Silver PA, Collins JJ, Yin P (2014) Toehold Switches: De-Novo-Designed Regulators of Gene Expression. Cell, 159, 925–939. https://doi.org/10.1016/j.cell.2014.10.002 Kavita K, Breaker RR (2022) Discovering riboswitches: the past and the future. Trends in Biochemical Sciences. https://doi.org/10.1016/j.tibs.2022.08.009 Tickner ZJ, Farzan M (2021) Riboswitches for Controlled Expression of Therapeutic Transgenes Delivered by Adeno-Associated Viral Vectors. Pharmaceuticals, 14, 554. https://doi.org/10.3390/ph14060554 To AC-Y, Chu DH-T, Wang AR, Li FC-Y, Chiu AW-O, Gao DY, Choi CHJ, Kong S-K, Chan T-F, Chan K-M, Yip KY (2018) A comprehensive web tool for toehold switch design. Bioinformatics, 34, 2862–2864. https://doi.org/10.1093/bioinformatics/bty216 | Toeholder: a Software for Automated Design and In Silico Validation of Toehold Riboswitches | Angel F. Cisneros, François D. Rouleau, Carla Bautista, Pascale Lemieux, Nathan Dumont-Leblond | <p>Abstract: Synthetic biology aims to engineer biological circuits, which often involve gene expression. A particularly promising group of regulatory elements are riboswitches because of their versatility with respect to their targets, but e... | | Bioinformatics | Sahar Melamed | 2022-02-16 14:40:13 | ||
22 Nov 2023
The slow evolving genome of the xenacoelomorph worm Xenoturbella bockiGenomic idiosyncrasies of Xenoturbella bocki: morphologically simple yet genetically complexRecommended by Rosa Fernández based on reviews by Christopher Laumer and 1 anonymous reviewerXenoturbella is a genus of morphologically simple bilaterians inhabiting benthic environments. Until very recently, only one species was known from the genus, Xenoturbella bocki Westblad 1949 [1]. Less than a decade ago, five more species were discovered (X. churro, X. monstrosa, X. profunda, X. hollandorum [2] and X. japonica [3]). These enigmatic animals lack an anus, a coelom, reproductive organs, nephrocytes and a centralized nervous system [1]. The systematic classification of the genus has substantially changed in the last decades, with first being considered as its own phylum (Xenoturbellida) and then being clustered together with acoels and nemertodermatids into the phylum Xenacoelomorpha [4,5]. The phylogenetic position of the xenacoelomorphs has been recalcitrant to resolution, with its position ranging from being the sister group to Nephrozoa (ie, protostomes and deuterostomes [6]) to the sister group to Ambulacraria (ie, Hemichordata and Echinodermata) in a clade called Xenambulacraria [4]. Recent studies based on expanded datasets and more refined analyses support either topology [7,8]. Either way, it is clear that additional studies on Xenoturbella could provide important insights into the origins of bilaterian traits such as the anus, the nephrons and the evolution of a centralized nervous system.
In any case, we are approaching a qualitative jump in how we understand phylogenomics thanks to efforts derived from the availability of chromosome-level genome assemblies for a growing number of species. Exciting times are ahead for us, evolutionary biologists, to explore what high-quality genomes - in combination with multiomics datasets - will reveal about animal evolution. I am personally really looking forward to it. References 1. Westblad E. (1949). Xenoturbella bocki n.g., n.sp., a peculiar, primitive Turbellarian type. Arkiv för Zoologi 1, 3-29 (1949). 2. Rouse, G. W., Wilson, N. G., Carvajal, J. I. & Vrijenhoek, R. C. New deep-sea species of Xenoturbella and the position of Xenacoelomorpha. Nature 530, 94–97 (2016). https://doi.org/10.1038/nature16545 3. Nakano, H. et al. Correction to: A new species of Xenoturbella from the western Pacific Ocean and the evolution of Xenoturbella. BMC Evol. Biol. 18, 1–2 (2018). https://doi.org/10.1186/s12862-018-1190-5https://doi.org/10.1186/s12862-018-1190-5 4. Philippe, H. et al. Acoelomorph flatworms are deuterostomes related to Xenoturbella. Nature 470, 255–258 (2011). https://doi.org/10.1038/nature09676 5. Hejnol, A. et al. Assessing the root of bilaterian animals with scalable phylogenomic methods. Proc. Biol. Sci. 276, 4261–4270 (2009). https://doi.org/10.1098/rspb.2009.0896 6. Cannon, J. T. et al. Xenacoelomorpha is the sister group to Nephrozoa. Nature 530, 89–93 (2016). https://doi.org/10.1038/nature16520 7. Laumer, C. E. et al. Revisiting metazoan phylogeny with genomic sampling of all phyla. Proc. Biol. Sci. 286, 20190831 (2019). https://doi.org/10.1098/rspb.2019.0831 8. Philippe, H. et al. Mitigating anticipated effects of systematic errors supports sister-group relationship between Xenacoelomorpha and Ambulacraria. Curr. Biol. 29, 1818–1826.e6 (2019). https://doi.org/10.1016/j.cub.2019.04.009 9. Schiffer, P. H., Natsidis, P., Leite D. J., Robertson, H., Lapraz, F., Marlétaz, F., Fromm, B., Baudry, L., Simpson, F., Høye, E., Zakrzewski, A-C., Kapli, P., Hoff, K. J., Mueller, S., Marbouty, M., Marlow, H., Copley, R. R., Koszul, R., Sarkies, P. & Telford, M .J. The slow evolving genome of the xenacoelomorph worm Xenoturbella bocki. bioRxiv (2023), ver. 4 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2022.06.24.497508 10. Suga, H. et al. The Capsaspora genome reveals a complex unicellular prehistory of animals. Nat. Commun. 4, 2325 (2013). https://doi.org/10.1038/ncomms3325 11. Fernández, R. & Gabaldón, T. Gene gain and loss across the metazoan tree of life. Nat Ecol Evol 4, 524–533 (2020). https://doi.org/10.1038/s41559-019-1069-x | The slow evolving genome of the xenacoelomorph worm *Xenoturbella bocki* | Philipp H. Schiffer, Paschalis Natsidis, Daniel J. Leite, Helen Robertson, François Lapraz, Ferdinand Marlétaz, Bastian Fromm, Liam Baudry, Fraser Simpson, Eirik Høye, Anne-C. Zakrzewski, Paschalia Kapli, Katharina J. Hoff, Steven Mueller, Martial... | <p style="text-align: justify;">The evolutionary origins of Bilateria remain enigmatic. One of the more enduring proposals highlights similarities between a cnidarian-like planula larva and simple acoel-like flatworms. This idea is based in part o... | | Evolutionary genomics | Rosa Fernández | 2022-11-01 12:31:53 | ||
06 Feb 2024

The need of decoding life for taking care of biodiversity and the sustainable use of nature in the Anthropocene - a Faroese perspectiveWhy sequence everything? A raison d’être for the Genome Atlas of Faroese EcologyRecommended by Stephen Richards based on reviews by Tereza Manousaki and 1 anonymous reviewer
When discussing the Earth BioGenome Project with scientists and potential funding agencies, one common question is: why sequence everything? Whether sequencing a subset would be more optimal is not an unreasonable question given what we know about the mathematics of importance and Pareto’s 80:20 principle, that 80% of the benefits can come from 20% of the effort. However, one must remember that this principle is an observation made in hindsight and selecting the most effective 20% of experiments is difficult. As an example, few saw great applied value in comparative genomic analysis of the archaea Haloferax mediterranei, but this enabled the discovery of CRISPR/Cas9 technology (1). When discussing whether or not to sequence all life on our planet, smaller countries such as the Faroe Islands are seldom mentioned.
1 Mojica, F. J., Díez-Villaseñor, C. S., García-Martínez, J. & Soria, E. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J Mol Evol 60, 174-182 (2005). 2 Mikalsen, S-O., Hjøllum, J. í., Salter, I., Djurhuus, A. & Kongsstovu, S. í. The need of decoding life for taking care of biodiversity and the sustainable use of nature in the Anthropocene – a Faroese perspective. EcoEvoRxiv (2024), ver. 3 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.32942/X21S4C | The need of decoding life for taking care of biodiversity and the sustainable use of nature in the Anthropocene - a Faroese perspective | Svein-Ole Mikalsen, Jari í Hjøllum, Ian Salter, Anni Djurhuus, Sunnvør í Kongsstovu | <p>Biodiversity is under pressure, mainly due to human activities and climate change. At the international policy level, it is now recognised that genetic diversity is an important part of biodiversity. The availability of high-quality reference g... | | ERGA, ERGA Pilot, Population genomics, Vertebrates | Stephen Richards | 2023-07-31 16:59:33 | ||
15 Jan 2024
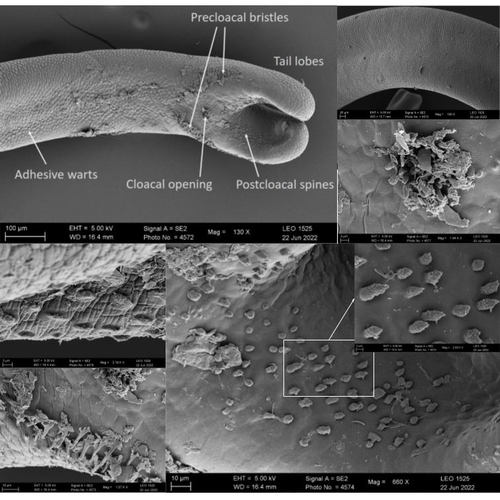
The genome sequence of the Montseny horsehair worm, Gordionus montsenyensis sp. nov., a key resource to investigate Ecdysozoa evolutionEmbarking on a novel journey in Metazoa evolution through the pioneering sequencing of a key underrepresented lineageRecommended by Juan C. Opazo based on reviews by Gonzalo Riadi and 2 anonymous reviewers
Whole genome sequences are revolutionizing our understanding across various biological fields. They not only shed light on the evolution of genetic material but also uncover the genetic basis of phenotypic diversity. The sequencing of underrepresented lineages, such as the one presented in this study, is of critical importance. It is crucial in filling significant gaps in our understanding of Metazoa evolution. Despite the wealth of genome sequences in public databases, it is crucial to acknowledge that some lineages across the Tree of Life are underrepresented or absent. This research represents a significant step towards addressing this imbalance, contributing to the collective knowledge of the global scientific community. In this genome note, as part of the European Reference Genome Atlas pilot effort to generate reference genomes for European biodiversity (Mc Cartney et al. 2023), Klara Eleftheriadi and colleagues (Eleftheriadi et al. 2023) make a significant effort to add a genome sequence of an unrepresented group in the animal Tree of Life. More specifically, they present a taxonomic description and chromosome-level genome assembly of a newly described species of horsehair worm (Gordionus montsenyensis). Their sequence methodology gave rise to an assembly of 396 scaffolds totaling 288 Mb, with an N50 value of 64.4 Mb, where 97% of this assembly is grouped into five pseudochromosomes. The nuclear genome annotation predicted 10,320 protein-coding genes, and they also assembled the circular mitochondrial genome into a 15-kilobase sequence. The selection of a species representing the phylum Nematomorpha, a group of parasitic organisms belonging to the Ecdysozoa lineage, is good, since today, there is only one publicly available genome for this animal phylum (Cunha et al. 2023). Interestingly, this article shows, among other things, that the species analyzed has lost ∼30% of the universal Metazoan genes. Efforts, like the one performed by Eleftheriadi and colleagues, are necessary to gain more insights, for example, on the evolution of this massive gene lost in this group of animals.
Cunha, T. J., de Medeiros, B. A. S, Lord, A., Sørensen, M. V., and Giribet, G. (2023). Rampant Loss of Universal Metazoan Genes Revealed by a Chromosome-Level Genome Assembly of the Parasitic Nematomorpha. Current Biology, 33 (16): 3514–21.e4. https://doi.org/10.1016/j.cub.2023.07.003 Eleftheriadi, K., Guiglielmoni, N., Salces-Ortiz, J., Vargas-Chavez, C., Martínez-Redondo, G. I., Gut, M., Flot, J.-F., Schmidt-Rhaesa, A., and Fernández, R. (2023). The Genome Sequence of the Montseny Horsehair worm, Gordionus montsenyensis sp. Nov., a Key Resource to Investigate Ecdysozoa Evolution. bioRxiv, ver. 3 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2023.06.26.546503 Mc Cartney, A. M., Formenti, G., Mouton, A., De Panis, D., Marins, L. S., Leitão, H. G., Diedericks, G., et al. (2023). The European Reference Genome Atlas: Piloting a Decentralised Approach to Equitable Biodiversity Genomics. bioRxiv. https://doi.org/10.1101/2023.09.25.559365 | The genome sequence of the Montseny horsehair worm, *Gordionus montsenyensis* sp. nov., a key resource to investigate Ecdysozoa evolution | Eleftheriadi Klara, Guiglielmoni Nadège, Salces-Ortiz Judit, Vargas-Chávez Carlos, Martínez-Redondo Gemma I, Gut Marta, Flot Jean François, Schmidt-Rhaesa Andreas, Fernández Rosa | <p>Nematomorpha, also known as Gordiacea or Gordian worms, are a phylum of parasitic organisms that belong to the Ecdysozoa, a clade of invertebrate animals characterized by molting. They are one of the less scientifically studied animal phyla, an... | | ERGA Pilot | Juan C. Opazo | 2023-06-29 10:31:36 | ||
07 Sep 2023
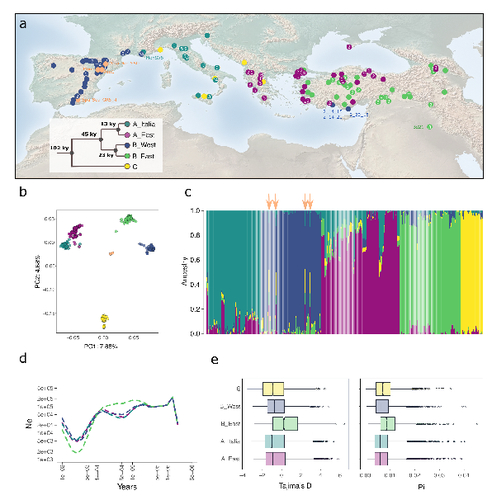
The demographic history of the wild crop relative Brachypodium distachyon is shaped by distinct past and present ecological nichesNatural variation and adaptation in Brachypodium distachyonRecommended by Josep Casacuberta based on reviews by Thibault Leroy and 1 anonymous reviewerIdentifying the genetic factors that allow plant adaptation is a major scientific question that is particularly relevant in the face of the climate change that we are already experiencing. To address this, it is essential to have genetic information on a high number of accessions (i.e., plants registered with unique accession numbers) growing under contrasting environmental conditions. There is already an important number of studies addressing these issues in the plant Arabidopsis thaliana, but there is a need to expand these analyses to species that play key roles in wild ecosystems and are close to very relevant crops, as is the case of grasses. The work of Minadakis, Roulin and co-workers (1) presents a Brachypodium distachyon panel of 332 fully sequences accessions that covers the whole species distribution across a wide range of bioclimatic conditions, which will be an invaluable tool to fill this gap. In addition, the authors use this data to start analyzing the population structure and demographic history of this plant, suggesting that the species experienced a shift of its distribution following the Last Glacial Maximum, which may have forced the species into new habitats. The authors also present a modeling of the niches occupied by B. distachyon together with an analysis of the genetic clades found in each of them, and start analyzing the different adaptive loci that may have allowed the species’ expansion into different bioclimatic areas. In addition to the importance of the resources made available by the authors for the scientific community, the analyses presented are well done and carefully discussed, and they highlight the potential of these new resources to investigate the genetic bases of plant adaptation. References 1. Nikolaos Minadakis, Hefin Williams, Robert Horvath, Danka Caković, Christoph Stritt, Michael Thieme, Yann Bourgeois, Anne C. Roulin. The demographic history of the wild crop relative Brachypodium distachyon is shaped by distinct past and present ecological niches. bioRxiv, 2023.06.01.543285, ver. 5 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2023.06.01.543285 | The demographic history of the wild crop relative *Brachypodium distachyon* is shaped by distinct past and present ecological niches | Nikolaos Minadakis, Hefin Williams, Robert Horvath, Danka Caković, Christoph Stritt, Michael Thieme, Yann Bourgeois, Anne C. Roulin | <p style="text-align: justify;">Closely related to economically important crops, the grass <em>Brachypodium distachyon</em> has been originally established as a pivotal species for grass genomics but more recently flourished as a model for develop... | | Evolutionary genomics, Functional genomics, Plants, Population genomics | Josep Casacuberta | 2023-06-14 15:28:30 | ||
08 Nov 2022

Somatic mutation detection: a critical evaluation through simulations and reanalyses in oaksHow to best call the somatic mosaic tree?Recommended by Nicolas Bierne based on reviews by 2 anonymous reviewersAny multicellular organism is a molecular mosaic with some somatic mutations accumulated between cell lineages. Big long-lived trees have nourished this imaginary of a somatic mosaic tree, from the observation of spectacular phenotypic mosaics and also because somatic mutations are expected to potentially be passed on to gametes in plants (review in Schoen and Schultz 2019). The lower cost of genome sequencing now offers the opportunity to tackle the issue and identify somatic mutations in trees. However, when it comes to characterizing this somatic mosaic from genome sequences, things become much more difficult than one would think in the first place. What separates cell lineages ontogenetically, in cell division number, or in time? How to sample clonal cell populations? How do somatic mutations distribute in a population of cells in an organ or an organ sample? Should they be fixed heterozygotes in the sample of cells sequenced or be polymorphic? Do we indeed expect somatic mutations to be fixed? How should we identify and count somatic mutations? To date, the detection of somatic mutations has mostly been done with a single variant caller in a given study, and we have little perspective on how different callers provide similar or different results. Some studies have used standard SNP callers that assumed a somatic mutation is fixed at the heterozygous state in the sample of cells, with an expected allele coverage ratio of 0.5, and less have used cancer callers, designed to detect mutations in a fraction of the cells in the sample. However, standard SNP callers detect mutations that deviate from a balanced allelic coverage, and different cancer callers can have different characteristics that should affect their outcomes. In order to tackle these issues, Schmitt et al. (2022) conducted an extensive simulation analysis to compare different variant callers. Then, they reanalyzed two large published datasets on pedunculate oak, Quercus robur. The analysis of in silico somatic mutations allowed the authors to evaluate the performance of different variant callers as a function of the allelic fraction of somatic mutations and the sequencing depth. They found one of the seven callers to provide better and more robust calls for a broad set of allelic fractions and sequencing depths. The reanalysis of published datasets in oaks with the most effective cancer caller of the in silico analysis allowed them to identify numerous low-frequency mutations that were missed in the original studies. I recommend the study of Schmitt et al. (2022) first because it shows the benefit of using cancer callers in the study of somatic mutations, whatever the allelic fraction you are interested in at the end. You can select fixed heterozygotes if this is your ultimate target, but cancer callers allow you to have in addition a valuable overview of the allelic fractions of somatic mutations in your sample, and most do as well as SNP callers for fixed heterozygous mutations. In addition, Schmitt et al. (2022) provide the pipelines that allow investigating in silico data that should correspond to a given study design, encouraging to compare different variant callers rather than arbitrarily going with only one. We can anticipate that the study of somatic mutations in non-model species will increasingly attract attention now that multiple tissues of the same individual can be sequenced at low cost, and the study of Schmitt et al. (2022) paves the way for questioning and choosing the best variant caller for the question one wants to address. References Schoen DJ, Schultz ST (2019) Somatic Mutation and Evolution in Plants. Annual Review of Ecology, Evolution, and Systematics, 50, 49–73. https://doi.org/10.1146/annurev-ecolsys-110218-024955 Schmitt S, Leroy T, Heuertz M, Tysklind N (2022) Somatic mutation detection: a critical evaluation through simulations and reanalyses in oaks. bioRxiv, 2021.10.11.462798. ver. 4 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2021.10.11.462798 | Somatic mutation detection: a critical evaluation through simulations and reanalyses in oaks | Sylvain Schmitt, Thibault Leroy, Myriam Heuertz, Niklas Tysklind | <p style="text-align: justify;">1. Mutation, the source of genetic diversity, is the raw material of evolution; however, the mutation process remains understudied, especially in plants. Using both a simulation and reanalysis framework, we set out ... | | Bioinformatics, Plants | Nicolas Bierne | Anonymous, Anonymous | 2022-04-28 13:24:19 | |
10 Jul 2023
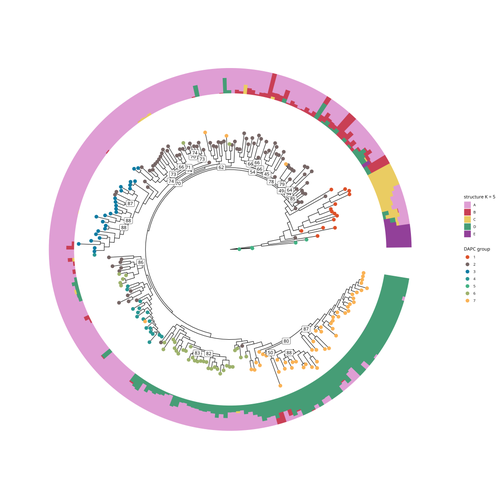
SNP discovery by exome capture and resequencing in a pea genetic resource collectionThe value of a large Pisum SNP datasetRecommended by Wanapinun Nawae based on reviews by Rui Borges and 1 anonymous reviewerOne important goal of modern genetics is to establish functional associations between genotype and phenotype. Single nucleotide polymorphisms (SNPs) are numerous and widely distributed in the genome and can be obtained from nucleic acid sequencing (1). SNPs allow for the investigation of genetic diversity, which is critical for increasing crop resilience to the challenges posed by global climate change. The associations between SNPs and phenotypes can be captured in genome-wide association studies. SNPs can also be used in combination with machine learning, which is becoming more popular for predicting complex phenotypic traits like yield and biotic and abiotic stress tolerance from genotypic data (2). The availability of many SNP datasets is important in machine learning predictions because this approach requires big data to build a comprehensive model of the association between genotype and phenotype. Aubert and colleagues have studied, as part of the PeaMUST project, the genetic diversity of 240 Pisum accessions (3). They sequenced exome-enriched genomic libraries, a technique that enables the identification of high-density, high-quality SNPs at a low cost (4). This technique involves capturing and sequencing only the exonic regions of the genome, which are the protein-coding regions. A total of 2,285,342 SNPs were obtained in this study. The analysis of these SNPs with the annotations of the genome sequence of one of the studied pea accessions (5) identified a number of SNPs that could have an impact on gene activity. Additional analyses revealed 647,220 SNPs that were unique to individual pea accessions, which might contribute to the fitness and diversity of accessions in different habitats. Phylogenetic and clustering analyses demonstrated that the SNPs could distinguish Pisum germplasms based on their agronomic and evolutionary histories. These results point out the power of selected SNPs as markers for identifying Pisum individuals. Overall, this study found high-quality SNPs that are meaningful in a biological context. This dataset was derived from a large set of germplasm and is thus particularly useful for studying genotype-phenotype associations, as well as the diversity within Pisum species. These SNPs could also be used in breeding programs to develop new pea varieties that are resilient to abiotic and biotic stressors. References
https://doi.org/10.1139/gen-2021-005
https://doi.org/10.1186/s12870-022-03559-z
https://doi.org/10.1101/2022.08.03.502586
https://doi.org/10.1534/g3.115.018564
| SNP discovery by exome capture and resequencing in a pea genetic resource collection | G. Aubert, J. Kreplak, M. Leveugle, H. Duborjal, A. Klein, K. Boucherot, E. Vieille, M. Chabert-Martinello, C. Cruaud, V. Bourion, I. Lejeune-Hénaut, M.L. Pilet-Nayel, Y. Bouchenak-Khelladi, N. Francillonne, N. Tayeh, J.P. Pichon, N. Rivière, J. B... | <p style="text-align: justify;"><strong>Background & Summary</strong></p> <p style="text-align: justify;">In addition to being the model plant used by Mendel to establish genetic laws, pea (<em>Pisum sativum</em> L., 2n=14) is a major pulse c... | | Plants, Population genomics | Wanapinun Nawae | 2022-11-29 09:29:06 |




