
Latest recommendations

| Id | Title | Authors | Abstract | Picture | Thematic fields▼ | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
10 Jul 2023
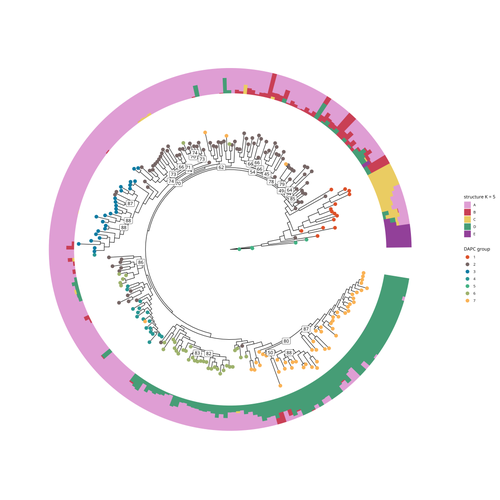
SNP discovery by exome capture and resequencing in a pea genetic resource collectionThe value of a large Pisum SNP datasetRecommended by Wanapinun Nawae based on reviews by Rui Borges and 1 anonymous reviewerOne important goal of modern genetics is to establish functional associations between genotype and phenotype. Single nucleotide polymorphisms (SNPs) are numerous and widely distributed in the genome and can be obtained from nucleic acid sequencing (1). SNPs allow for the investigation of genetic diversity, which is critical for increasing crop resilience to the challenges posed by global climate change. The associations between SNPs and phenotypes can be captured in genome-wide association studies. SNPs can also be used in combination with machine learning, which is becoming more popular for predicting complex phenotypic traits like yield and biotic and abiotic stress tolerance from genotypic data (2). The availability of many SNP datasets is important in machine learning predictions because this approach requires big data to build a comprehensive model of the association between genotype and phenotype. Aubert and colleagues have studied, as part of the PeaMUST project, the genetic diversity of 240 Pisum accessions (3). They sequenced exome-enriched genomic libraries, a technique that enables the identification of high-density, high-quality SNPs at a low cost (4). This technique involves capturing and sequencing only the exonic regions of the genome, which are the protein-coding regions. A total of 2,285,342 SNPs were obtained in this study. The analysis of these SNPs with the annotations of the genome sequence of one of the studied pea accessions (5) identified a number of SNPs that could have an impact on gene activity. Additional analyses revealed 647,220 SNPs that were unique to individual pea accessions, which might contribute to the fitness and diversity of accessions in different habitats. Phylogenetic and clustering analyses demonstrated that the SNPs could distinguish Pisum germplasms based on their agronomic and evolutionary histories. These results point out the power of selected SNPs as markers for identifying Pisum individuals. Overall, this study found high-quality SNPs that are meaningful in a biological context. This dataset was derived from a large set of germplasm and is thus particularly useful for studying genotype-phenotype associations, as well as the diversity within Pisum species. These SNPs could also be used in breeding programs to develop new pea varieties that are resilient to abiotic and biotic stressors. References
https://doi.org/10.1139/gen-2021-005
https://doi.org/10.1186/s12870-022-03559-z
https://doi.org/10.1101/2022.08.03.502586
https://doi.org/10.1534/g3.115.018564
| SNP discovery by exome capture and resequencing in a pea genetic resource collection | G. Aubert, J. Kreplak, M. Leveugle, H. Duborjal, A. Klein, K. Boucherot, E. Vieille, M. Chabert-Martinello, C. Cruaud, V. Bourion, I. Lejeune-Hénaut, M.L. Pilet-Nayel, Y. Bouchenak-Khelladi, N. Francillonne, N. Tayeh, J.P. Pichon, N. Rivière, J. B... | <p style="text-align: justify;"><strong>Background & Summary</strong></p> <p style="text-align: justify;">In addition to being the model plant used by Mendel to establish genetic laws, pea (<em>Pisum sativum</em> L., 2n=14) is a major pulse c... | | Plants, Population genomics | Wanapinun Nawae | 2022-11-29 09:29:06 | ||
15 Dec 2022

Botrytis cinerea strains infecting grapevine and tomato display contrasted repertoires of accessory chromosomes, transposons and small RNAsExploring genomic determinants of host specialization in Botrytis cinereaRecommended by Sebastien Duplessis based on reviews by Cecile Lorrain and Thorsten LangnerThe genomics era has pushed forward our understanding of fungal biology. Much progress has been made in unraveling new gene functions and pathways, as well as the evolution or adaptation of fungi to their hosts or environments through population studies (Hartmann et al. 2019; Gladieux et al. 2018). Closing gaps more systematically in draft genomes using the most recent long-read technologies now seems the new standard, even with fungal species presenting complex genome structures (e.g. large and highly repetitive dikaryotic genomes; Duan et al. 2022). Understanding the genomic dynamics underlying host specialization in phytopathogenic fungi is of utmost importance as it may open new avenues to combat diseases. A strong host specialization is commonly observed for biotrophic and hemi-biotrophic fungal species or for necrotrophic fungi with a narrow host range, whereas necrotrophic fungi with broad host range are considered generalists (Liang and Rollins, 2018; Newman and Derbyshire, 2020). However, some degrees of specialization towards given hosts have been reported in generalist fungi and the underlying mechanisms remain to be determined. Botrytis cinerea is a polyphagous necrotrophic phytopathogen with a particularly wide host range and it is notably responsible for grey mould disease on many fruits, such as tomato and grapevine. Because of its importance as a plant pathogen, its relatively small genome size and its taxonomical position, it has been targeted for early genome sequencing and a first reference genome was provided in 2011 (Amselem et al. 2011). Other genomes were subsequently sequenced for other strains, and most importantly a gapless assembled version of the initial reference genome B05.10 was provided to the community (van Kan et al. 2017). This genomic resource has supported advances in various aspects of the biology of B. cinerea such as the production of specialized metabolites, which plays an important role in host-plant colonization, or more recently in the production of small RNAs which interfere with the host immune system, representing a new class of non-proteinaceous virulence effectors (Dalmais et al. 2011; Weiberg et al. 2013). In the present study, Simon et al. (2022) use PacBio long-read sequencing for Sl3 and Vv3 strains, which represent genetic clusters in B. cinerea populations found on tomato and grapevine. The authors combined these complete and high-quality genome assemblies with the B05.10 reference genome and population sequencing data to perform a comparative genomic analysis of specialization towards the two host plants. Transposable elements generate genomic diversity due to their mobile and repetitive nature and they are of utmost importance in the evolution of fungi as they deeply reshape the genomic landscape (Lorrain et al. 2021). Accessory chromosomes are also known drivers of adaptation in fungi (Möller and Stukenbrock, 2017). Here, the authors identify several genomic features such as the presence of different sets of accessory chromosomes, the presence of differentiated repertoires of transposable elements, as well as related small RNAs in the tomato and grapevine populations, all of which may be involved in host specialization. Whereas core chromosomes are highly syntenic between strains, an accessory chromosome validated by pulse-field electrophoresis is specific of the strains isolated from grapevine. Particularly, they show that two particular retrotransposons are discriminant between the strains and that they allow the production of small RNAs that may act as effectors. The discriminant accessory chromosome of the Vv3 strain harbors one of the unraveled retrotransposons as well as new genes of yet unidentified function. I recommend this article because it perfectly illustrates how efforts put into generating reference genomic sequences of higher quality can lead to new discoveries and allow to build strong hypotheses about biology and evolution in fungi. Also, the study combines an up-to-date genomics approach with a classical methodology such as pulse-field electrophoresis to validate the presence of accessory chromosomes. A major input of this investigation of the genomic determinants of B. cinerea is that it provides solid hints for further analysis of host-specialization at the population level in a broad-scale phytopathogenic fungus. References Amselem J, Cuomo CA, Kan JAL van, Viaud M, Benito EP, Couloux A, Coutinho PM, Vries RP de, Dyer PS, Fillinger S, Fournier E, Gout L, Hahn M, Kohn L, Lapalu N, Plummer KM, Pradier J-M, Quévillon E, Sharon A, Simon A, Have A ten, Tudzynski B, Tudzynski P, Wincker P, Andrew M, Anthouard V, Beever RE, Beffa R, Benoit I, Bouzid O, Brault B, Chen Z, Choquer M, Collémare J, Cotton P, Danchin EG, Silva CD, Gautier A, Giraud C, Giraud T, Gonzalez C, Grossetete S, Güldener U, Henrissat B, Howlett BJ, Kodira C, Kretschmer M, Lappartient A, Leroch M, Levis C, Mauceli E, Neuvéglise C, Oeser B, Pearson M, Poulain J, Poussereau N, Quesneville H, Rascle C, Schumacher J, Ségurens B, Sexton A, Silva E, Sirven C, Soanes DM, Talbot NJ, Templeton M, Yandava C, Yarden O, Zeng Q, Rollins JA, Lebrun M-H, Dickman M (2011) Genomic Analysis of the Necrotrophic Fungal Pathogens Sclerotinia sclerotiorum and Botrytis cinerea. PLOS Genetics, 7, e1002230. https://doi.org/10.1371/journal.pgen.1002230 Dalmais B, Schumacher J, Moraga J, Le Pêcheur P, Tudzynski B, Collado IG, Viaud M (2011) The Botrytis cinerea phytotoxin botcinic acid requires two polyketide synthases for production and has a redundant role in virulence with botrydial. Molecular Plant Pathology, 12, 564–579. https://doi.org/10.1111/j.1364-3703.2010.00692.x Duan H, Jones AW, Hewitt T, Mackenzie A, Hu Y, Sharp A, Lewis D, Mago R, Upadhyaya NM, Rathjen JP, Stone EA, Schwessinger B, Figueroa M, Dodds PN, Periyannan S, Sperschneider J (2022) Physical separation of haplotypes in dikaryons allows benchmarking of phasing accuracy in Nanopore and HiFi assemblies with Hi-C data. Genome Biology, 23, 84. https://doi.org/10.1186/s13059-022-02658-2 Gladieux P, Condon B, Ravel S, Soanes D, Maciel JLN, Nhani A, Chen L, Terauchi R, Lebrun M-H, Tharreau D, Mitchell T, Pedley KF, Valent B, Talbot NJ, Farman M, Fournier E (2018) Gene Flow between Divergent Cereal- and Grass-Specific Lineages of the Rice Blast Fungus Magnaporthe oryzae. mBio, 9, e01219-17. https://doi.org/10.1128/mBio.01219-17 Hartmann FE, Rodríguez de la Vega RC, Carpentier F, Gladieux P, Cornille A, Hood ME, Giraud T (2019) Understanding Adaptation, Coevolution, Host Specialization, and Mating System in Castrating Anther-Smut Fungi by Combining Population and Comparative Genomics. Annual Review of Phytopathology, 57, 431–457. https://doi.org/10.1146/annurev-phyto-082718-095947 Liang X, Rollins JA (2018) Mechanisms of Broad Host Range Necrotrophic Pathogenesis in Sclerotinia sclerotiorum. Phytopathology®, 108, 1128–1140. https://doi.org/10.1094/PHYTO-06-18-0197-RVW Lorrain C, Oggenfuss U, Croll D, Duplessis S, Stukenbrock E (2021) Transposable Elements in Fungi: Coevolution With the Host Genome Shapes, Genome Architecture, Plasticity and Adaptation. In: Encyclopedia of Mycology (eds Zaragoza Ó, Casadevall A), pp. 142–155. Elsevier, Oxford. https://doi.org/10.1016/B978-0-12-819990-9.00042-1 Möller M, Stukenbrock EH (2017) Evolution and genome architecture in fungal plant pathogens. Nature Reviews Microbiology, 15, 756–771. https://doi.org/10.1038/nrmicro.2017.76 Newman TE, Derbyshire MC (2020) The Evolutionary and Molecular Features of Broad Host-Range Necrotrophy in Plant Pathogenic Fungi. Frontiers in Plant Science, 11. https://doi.org/10.3389/fpls.2020.591733 Simon A, Mercier A, Gladieux P, Poinssot B, Walker A-S, Viaud M (2022) Botrytis cinerea strains infecting grapevine and tomato display contrasted repertoires of accessory chromosomes, transposons and small RNAs. bioRxiv, 2022.03.07.483234, ver. 4 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2022.03.07.483234 Van Kan JAL, Stassen JHM, Mosbach A, Van Der Lee TAJ, Faino L, Farmer AD, Papasotiriou DG, Zhou S, Seidl MF, Cottam E, Edel D, Hahn M, Schwartz DC, Dietrich RA, Widdison S, Scalliet G (2017) A gapless genome sequence of the fungus Botrytis cinerea. Molecular Plant Pathology, 18, 75–89. https://doi.org/10.1111/mpp.12384 Weiberg A, Wang M, Lin F-M, Zhao H, Zhang Z, Kaloshian I, Huang H-D, Jin H (2013) Fungal Small RNAs Suppress Plant Immunity by Hijacking Host RNA Interference Pathways. Science, 342, 118–123. https://doi.org/10.1126/science.1239705 | Botrytis cinerea strains infecting grapevine and tomato display contrasted repertoires of accessory chromosomes, transposons and small RNAs | Adeline Simon, Alex Mercier, Pierre Gladieux, Benoit Poinssot, Anne-Sophie Walker, Muriel Viaud | <p style="text-align: justify;">The fungus <em>Botrytis cinerea</em> is a polyphagous pathogen that encompasses multiple host-specialized lineages. While several secreted proteins, secondary metabolites and retrotransposons-derived small RNAs have... | | Fungi, Structural genomics, Viruses and transposable elements | Sebastien Duplessis | Cecile Lorrain, Thorsten Langner | 2022-03-15 11:15:48 | |
25 Nov 2022
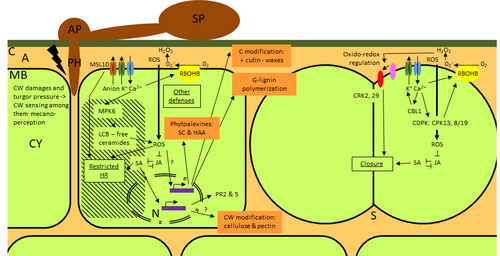
Phenotypic and transcriptomic analyses reveal major differences between apple and pear scab nonhost resistanceApples and pears: two closely related species with differences in scab nonhost resistanceRecommended by Wirulda Pootakham based on reviews by 3 anonymous reviewersNonhost resistance is a common form of disease resistance exhibited by plants against microorganisms that are pathogenic to other plant species [1]. Apples and pears are two closely related species belonging to Rosaceae family, both affected by scab disease caused by fungal pathogens in the Venturia genus. These pathogens appear to be highly host-specific. While apples are nonhosts for Venturia pyrina, pears are nonhosts for Venturia inaequalis. To date, the molecular bases of scab nonhost resistance in apple and pear have not been elucidated. This preprint by Vergne, et al (2022) [2] analyzed nonhost resistance symptoms in apple/V. pyrina and pear/V. inaequalis interactions as well as their transcriptomic responses. Interestingly, the author demonstrated that the nonhost apple/V. pyrina interaction was almost symptomless while hypersensitive reactions were observed for pear/V. inaequalis interaction. The transcriptomic analyses also revealed a number of differentially expressed genes (DEGs) that corresponded to the severity of the interactions, with very few DEGs observed during the apple/V. pyrina interaction and a much higher number of DEGs during the pear/V. inaequalis interaction. This type of reciprocal host-pathogen interaction study is valuable in gaining new insights into how plants interact with microorganisms that are potential pathogens in related species. A few processes appeared to be involved in the pear resistance against the nonhost pathogen V. inaequalis at the transcriptomic level, such as stomata closure, modification of cell wall and production of secondary metabolites as well as phenylpropanoids. Based on the transcriptomics changes during the nonhost interaction, the author compared the responses to those of host-pathogen interactions and revealed some interesting findings. They proposed a series of cascading effects in pear induced by the presence of V. inaequalis, which I believe helps shed some light on the basic mechanism for nonhost resistance. I am recommending this study because it provides valuable information that will strengthen our understanding of nonhost resistance in the Rosaceae family and other plant species. The knowledge gained here may be applied to genetically engineer plants for a broader resistance against a number of pathogens in the future. References 1. Senthil-Kumar M, Mysore KS (2013) Nonhost Resistance Against Bacterial Pathogens: Retrospectives and Prospects. Annual Review of Phytopathology, 51, 407–427. https://doi.org/10.1146/annurev-phyto-082712-102319 2. Vergne E, Chevreau E, Ravon E, Gaillard S, Pelletier S, Bahut M, Perchepied L (2022) Phenotypic and transcriptomic analyses reveal major differences between apple and pear scab nonhost resistance. bioRxiv, 2021.06.01.446506, ver. 4 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2021.06.01.446506 | Phenotypic and transcriptomic analyses reveal major differences between apple and pear scab nonhost resistance | E. Vergne, E. Chevreau, E. Ravon, S. Gaillard, S. Pelletier, M. Bahut, L. Perchepied | <p style="text-align: justify;"><strong>Background. </strong>Nonhost resistance is the outcome of most plant/pathogen interactions, but it has rarely been described in Rosaceous fruit species. Apple (<em>Malus x domestica</em> Borkh.) have a nonho... | | Functional genomics, Plants | Wirulda Pootakham | Jessica Soyer, Anonymous | 2022-05-13 15:06:08 | |
22 May 2023

Genetic bases of resistance to the rice hoja blanca disease deciphered by a QTL approachScoring symptoms of a plant viral diseaseRecommended by Olivier Panaud based on reviews by Grégoire Aubert and Valérie GeffroyThe paper from Silva et al. (2023) provides new insights into the genetic bases of natural resistance of rice to the Rice Hoja Blanca (RHB) disease, one of its most serious diseases in tropical countries of the American continent and the Caribbean. This disease is caused by the Rice Hoja Blanca Virus, or RHBV, the vector of which is the planthopper insect Tagosodes orizicolus Müir. It is responsible for serious damage to the rice crop (Morales and Jennings 2010). The authors take a Quantitative Trait Loci (QTL) detection approach to find genomic regions statistically associated with the resistant phenotype. To this aim, they use four resistant x susceptible crosses (the susceptible parent being the same in all four crosses) to maximize the chances to find new QTLs. The F2 populations derived from the crosses are genotyped using Single Nucleotide Polymorphisms (SNPs) extracted from whole-genome sequencing (WGS) data of the resistant parents, and the F3 families derived from the F2 individuals are scored for disease symptoms. For this, they use a computer-aided image analysis protocol that they designed so they can estimate the severity of the damages in the plant. They find several new QTLs, some being apparently more associated with disease severity, others with disease incidence. They also find that a previously identified QTL of Oryza sativa ssp. japonica origin is also present in the indica cluster (Romero et al. 2014). Finally, they discuss the candidate genes that could underlie the QTLs and provide a simple model for resistance. It has to be noted that scoring symptoms of a viral disease such as RHB is very challenging. It requires maintaining populations of viruliferous insect vectors, mastering times and conditions for infestation by nymphs, and precise symptom scoring. It also requires the preparation of segregating populations, their genotyping with enough genetic markers, and mastering QTL detection methods. All these aspects are present in this work. In particular, the phenotyping of symptom severity implemented using computer-aided image processing represents an impressive, enormous amount of work. From the genomics side, the fine-scale genotyping is based on the WGS of the parental lines (resistant and susceptible), followed by the application of suitable bioinformatic tools for SNP extraction and primers prediction that can be used on their Fluidigm platform. It also required implementing data correction algorithms to achieve precise genetic maps in the four crosses. The QTL detection itself required careful statistical pre-processing of phenotypic data. The authors then used a combination of several QTL detection methods, including an original meta-QTL method they developed in the software MapDisto. The authors then perform a very complete and convincing analysis of candidate genes, which includes genes already identified for a similar disease (RSV) on chromosome 11 of rice. What remains to elucidate is whether the candidate genes are actually involved or not in the disease resistance process. The team has already started implementing gene knockout strategies to study some of them in more detail. It will be interesting to see whether those genes act against the virus itself, or against the insect vector. Overall the work is of high quality and represents an important advance in the knowledge of disease resistance. In addition, it has many implications for crop breeding, allowing the setup of large-scale, marker-assisted strategies, for new resistant elite varieties of rice. References Morales F and Jennings P (2010) Rice hoja blanca: a complex plant-virus-vector pathosystem. CAB Reviews. https://doi.org/10.1079/PAVSNNR20105043 Romero LE, Lozano I, Garavito A, et al (2014) Major QTLs control resistance to Rice hoja blanca virus and its vector Tagosodes orizicolus. G3 | Genes, Genomes, Genetics 4:133–142. https://doi.org/10.1534/g3.113.009373 Silva A, Montoya ME, Quintero C, Cuasquer J, Tohme J, Graterol E, Cruz M, Lorieux M (2023) Genetic bases of resistance to the rice hoja blanca disease deciphered by a QTL approach. bioRxiv, 2022.11.07.515427, ver. 2 peer-reviewed and recommended by Peer Community in Genomics https://doi.org/10.1101/2022.11.07.515427 | Genetic bases of resistance to the rice hoja blanca disease deciphered by a QTL approach | Alexander Silva, Maria Elker Montoya, Constanza Quintero, Juan Cuasquer, Joe Tohme, Eduardo Graterol, Maribel Cruz, Mathias Lorieux | <p style="text-align: justify;">Rice hoja blanca (RHB) is one of the most serious diseases in rice growing areas in tropical Americas. Its causal agent is Rice hoja blanca virus (RHBV), transmitted by the planthopper <em>Tagosodes orizicolus </em>... | | Functional genomics, Plants | Olivier Panaud | 2022-11-09 09:13:30 | ||
07 Aug 2023
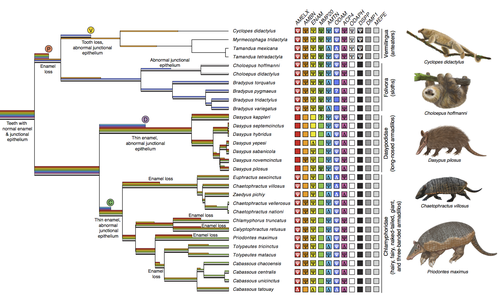
Genomic data suggest parallel dental vestigialization within the xenarthran radiationWhat does dental gene decay tell us about the regressive evolution of teeth in South American mammals?Recommended by Didier Casane based on reviews by Juan C. Opazo, Régis Debruyne and Nicolas PolletA group of mammals, Xenathra, evolved and diversified in South America during its long period of isolation in the early to mid Cenozoic era. More recently, as a result of the Great Faunal Interchange between South America and North America, many xenarthran species went extinct. The thirty-one extant species belong to three groups: armadillos, sloths and anteaters. They share dental degeneration. However, the level of degeneration is variable. Anteaters entirely lack teeth, sloths have intermediately regressed teeth and most armadillos have a toothless premaxilla, as well as peg-like, single-rooted teeth that lack enamel in adult animals (Vizcaíno 2009). This diversity raises a number of questions about the evolution of dentition in these mammals. Unfortunately, the fossil record is too poor to provide refined information on the different stages of regressive evolution in these clades. In such cases, the identification of loss-of-function mutations and/or relaxed selection in genes related to a character regression can be very informative (Emerling and Springer 2014; Meredith et al. 2014; Policarpo et al. 2021). Indeed, shared and unique pseudogenes/relaxed selection can tell us to what extent regression has occurred in common ancestors and whether some changes are lineage-specific. In addition, the distribution of pseudogenes/relaxed selection on the branches of a phylogenetic tree is related to the evolutionary processes involved. A much higher density of pseudogenes in the most internal branches indicates that degeneration took place early and over a short period of time, consistent with selection against the presence of the morphological character with which they are associated, while pseudogenes distributed evenly in many internal and external branches suggest a more gradual process over many millions of years, in line with relaxed selection and fixation of loss-of-function mutations by genetic drift. In this paper (Emerling et al. 2023), the authors examined the dynamics of decay of 11 dental genes that may parallel teeth regression. The analyses of the data reported in this paper clearly point to xenarthran teeth having repeatedly regressed in parallel in the three clades. In fact, no loss-of-function mutation is shared by all species examined. However, more genes should be studied to confirm the hypothesis that the common ancestor of extant xenarthrans had normal dentition. There are distinct patterns of gene loss in different lineages that are associated with the variation in dentition observed across the clades. These patterns of gene loss suggest that regressive evolution took place both gradually and in relatively rapid, discrete phases during the diversification of xenarthrans. This study underscores the utility of using pseudogenes to reconstruct evolutionary history of morphological characters when fossils are sparse. References Emerling CA, Gibb GC, Tilak M-K, Hughes JJ, Kuch M, Duggan AT, Poinar HN, Nachman MW, Delsuc F. 2023. Genomic data suggest parallel dental vestigialization within the xenarthran radiation. bioRxiv, 2022.12.09.519446, ver 2, peer-reviewed and recommended by PCI Genomics. https://doi.org/10.1101/2022.12.09.519446 Emerling CA, Springer MS. 2014. Eyes underground: Regression of visual protein networks in subterranean mammals. Molecular Phylogenetics and Evolution 78: 260-270. https://doi.org/10.1016/j.ympev.2014.05.016 Meredith RW, Zhang G, Gilbert MTP, Jarvis ED, Springer MS. 2014. Evidence for a single loss of mineralized teeth in the common avian ancestor. Science 346: 1254390. https://doi.org/10.1126/science.1254390 Policarpo M, Fumey J, Lafargeas P, Naquin D, Thermes C, Naville M, Dechaud C, Volff J-N, Cabau C, Klopp C, et al. 2021. Contrasting gene decay in subterranean vertebrates: insights from cavefishes and fossorial mammals. Molecular Biology and Evolution 38: 589-605. https://doi.org/10.1093/molbev/msaa249 Vizcaíno SF. 2009. The teeth of the “toothless”: novelties and key innovations in the evolution of xenarthrans (Mammalia, Xenarthra). Paleobiology 35: 343-366. https://doi.org/10.1666/0094-8373-35.3.343 | Genomic data suggest parallel dental vestigialization within the xenarthran radiation | Christopher A Emerling, Gillian C Gibb, Marie-Ka Tilak, Jonathan J Hughes, Melanie Kuch, Ana T Duggan, Hendrik N Poinar, Michael W Nachman, Frederic Delsuc | <p style="text-align: justify;">The recent influx of genomic data has provided greater insights into the molecular basis for regressive evolution, or vestigialization, through gene loss and pseudogenization. As such, the analysis of gene degradati... | | Evolutionary genomics, Vertebrates | Didier Casane | 2022-12-12 16:01:57 | ||
07 Oct 2021

Fine-scale quantification of GC-biased gene conversion intensity in mammalsA systematic approach to the study of GC-biased gene conversion in mammalsRecommended by Carina Farah Mugal based on reviews by Fanny Pouyet , David Castellano and 1 anonymous reviewerThe role of GC-biased gene conversion (gBGC) in molecular evolution has interested scientists for the last two decades since its discovery in 1999 (Eyre-Walker 1999; Galtier et al. 2001). gBGC is a process that is associated with meiotic recombination, and is characterized by a transmission distortion in favor of G and C over A and T alleles at GC/AT heterozygous sites that occur in the vicinity of recombination-inducing double-strand breaks (Duret and Galtier 2009; Mugal et al. 2015). This transmission distortion results in a fixation bias of G and C alleles, equivalent to directional selection for G and C (Nagylaki 1983). The fixation bias subsequently leads to a correlation between recombination rate and GC content across the genome, which has served as indirect evidence for the prevalence of gBGC in many organisms. The fixation bias also produces shifts in the allele frequency spectrum (AFS) towards higher frequencies of G and C alleles. These molecular signatures of gBGC provide a means to quantify the strength of gBGC and study its variation among species and across the genome. Following this idea, first Lartillot (2013) and Capra et al. (2013) developed phylogenetic methodology to quantify gBGC based on substitutions, and De Maio et al. (2013) combined information on polymorphism into a phylogenetic setting. Complementary to the phylogenetic methods, later Glemin et al. (2015) developed a method that draws information solely from polymorphism data and the shape of the AFS. Application of these methods to primates (Capra et al. 2013; De Maio et al. 2013; Glemin et al. 2015) and mammals (Lartillot 2013) supported the notion that variation in the strength of gBGC across the genome reflects the dynamics of the recombination landscape, while variation among species correlates with proxies of the effective population size. However, application of the polymorphism-based method by Glemin et al. (2015) to distantly related Metazoa did not confirm the correlation with effective population size (Galtier et al. 2018). Here, Galtier (2021) introduces a novel phylogenetic approach applicable to the study of closely related species. Specifically, Galtier introduces a statistical framework that enables the systematic study of variation in the strength of gBGC among species and among genes. In addition, Galtier assesses fine-scale variation of gBGC across the genome by means of spatial autocorrelation analysis. This puts Galtier in a position to study variation in the strength of gBGC at three different scales, i) among species, ii) among genes, and iii) within genes. Galtier applies his method to four families of mammals, Hominidae, Cercopithecidae, Bovidae, and Muridae and provides a thorough discussion of his findings and methodology. Galtier found that the strength of gBGC correlates with proxies of the effective population size (Ne), but that the slope of the relationship differs among the four families of mammals. Given the relationship between the population-scaled strength of gBGC B = 4Neb, this finding suggests that the conversion bias (b) could vary among mammalian species. Variation in b could either result from differences in the strength of the transmission distortion (Galtier et al. 2018) or evolutionary changes in the rate of recombination (Boman et al. 2021). Alternatively, Galtier suggests that also systematic variation in proxies of Ne could lead to similar observations. Finally, the present study reports intriguing inter-species differences between the extent of variation in the strength of gBGC among and within genes, which are interpreted in consideration of the recombination dynamics in mammals. References Boman J, Mugal CF, Backström N (2021) The Effects of GC-Biased Gene Conversion on Patterns of Genetic Diversity among and across Butterfly Genomes. Genome Biology and Evolution, 13. https://doi.org/10.1093/gbe/evab064 Capra JA, Hubisz MJ, Kostka D, Pollard KS, Siepel A (2013) A Model-Based Analysis of GC-Biased Gene Conversion in the Human and Chimpanzee Genomes. PLOS Genetics, 9, e1003684. https://doi.org/10.1371/journal.pgen.1003684 De Maio N, Schlötterer C, Kosiol C (2013) Linking Great Apes Genome Evolution across Time Scales Using Polymorphism-Aware Phylogenetic Models. Molecular Biology and Evolution, 30, 2249–2262. https://doi.org/10.1093/molbev/mst131 Duret L, Galtier N (2009) Biased Gene Conversion and the Evolution of Mammalian Genomic Landscapes. Annual Review of Genomics and Human Genetics, 10, 285–311. https://doi.org/10.1146/annurev-genom-082908-150001 Eyre-Walker A (1999) Evidence of Selection on Silent Site Base Composition in Mammals: Potential Implications for the Evolution of Isochores and Junk DNA. Genetics, 152, 675–683. https://doi.org/10.1093/genetics/152.2.675 Galtier N (2021) Fine-scale quantification of GC-biased gene conversion intensity in mammals. bioRxiv, 2021.05.05.442789, ver. 5 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2021.05.05.442789 Galtier N, Piganeau G, Mouchiroud D, Duret L (2001) GC-Content Evolution in Mammalian Genomes: The Biased Gene Conversion Hypothesis. Genetics, 159, 907–911. https://doi.org/10.1093/genetics/159.2.907 Galtier N, Roux C, Rousselle M, Romiguier J, Figuet E, Glémin S, Bierne N, Duret L (2018) Codon Usage Bias in Animals: Disentangling the Effects of Natural Selection, Effective Population Size, and GC-Biased Gene Conversion. Molecular Biology and Evolution, 35, 1092–1103. https://doi.org/10.1093/molbev/msy015 Glémin S, Arndt PF, Messer PW, Petrov D, Galtier N, Duret L (2015) Quantification of GC-biased gene conversion in the human genome. Genome Research, 25, 1215–1228. https://doi.org/10.1101/gr.185488.114 Lartillot N (2013) Phylogenetic Patterns of GC-Biased Gene Conversion in Placental Mammals and the Evolutionary Dynamics of Recombination Landscapes. Molecular Biology and Evolution, 30, 489–502. https://doi.org/10.1093/molbev/mss239 Mugal CF, Weber CC, Ellegren H (2015) GC-biased gene conversion links the recombination landscape and demography to genomic base composition. BioEssays, 37, 1317–1326. https://doi.org/10.1002/bies.201500058 Nagylaki T (1983) Evolution of a finite population under gene conversion. Proceedings of the National Academy of Sciences, 80, 6278–6281. https://doi.org/10.1073/pnas.80.20.6278 | Fine-scale quantification of GC-biased gene conversion intensity in mammals | Nicolas Galtier | <p style="text-align: justify;">GC-biased gene conversion (gBGC) is a molecular evolutionary force that favours GC over AT alleles irrespective of their fitness effect. Quantifying the variation in time and across genomes of its intensity is key t... | | Evolutionary genomics, Population genomics, Vertebrates | Carina Farah Mugal | 2021-05-25 09:25:52 | ||
23 Aug 2022

A novel lineage of the Capra genus discovered in the Taurus Mountains of Turkey using ancient genomicsGoat ancient DNA analysis unveils a new lineage that may have hybridized with domestic goatsRecommended by Laura Botigué based on reviews by Torsten Günther and 1 anonymous reviewerThe genomic analysis of ancient remains has revolutionized the study of the past over the last decade. On top of the discoveries related to human evolution, plant and animal archaeogenomics has been used to gain new insights into the domestication process and the dispersal of domestic forms. In this study, Daly and colleagues analyse the genomic data from seven goat specimens from the Epipalaeolithic recovered from the Direkli Cave in the Taurus Mountains in southern Turkey. They also generate new genomic data from Capra lineages across the phylogeny, contributing to the availability of genomic resources for this genus. Analysis of the ancient remains is compared to modern genomic variability and sheds light on the complexity of the Tur wild Capra lineages and their relationship with domestic goats and their wild ancestors. Authors find that during the Late Pleistocene in the Taurus Mountains wild goats from the Tur lineage, today restricted to the Caucasus region, were not rare and cohabited with Bezoar, the wild goats that are the ancestors of domestic goats. They identify the Direkli Cave specimens as a lineage separate from the A modified D statistic, Dex, is developed to examine the contribution of the ancient Tur lineage in domestic goats through time and space. Dex measures the relative degree of allele sharing, derived specifically in a selected genome or group of genomes, and may have some utility in genera with complex admixture histories or admixture from ghost lineages. Results confirm that Neolithic European goat had an excess of allele sharing with this ancient Tur lineage, something that is absent in contemporary goats eastwards or in modern goats. Interspecific gene flow is not uncommon among mammals, but the case of Capra has the additional motivation of understanding the origins of the domestic species. This work uncovers an ancient Tur lineage that is different from the modern ones and is additionally found in another geographic area. Furthermore, evidence shows that this ancient lineage exhibits substantial amounts of allele sharing with the wild ancestor of the domestic goat, but also with the Neolithic Eurasian domestic goats, highlighting the complexity of the domestication process. This work has also important implications in understanding the effect of over-hunting and habitat disruption during the Anthropocene on the evolution of the Capra genus. The availability of more ancient specimens and better coverage of the modern genomic variability can help quantifying the lineages that went lost and identify the causes of their extinction. This work is limited by the current availability of whole genomes from modern Capra specimens, but pieces of evidence as well that an effort is needed to obtain more genomic data from ancient goats from different geographic ranges to determine to what extent these lineages contributed to goat domestication. References Daly KG, Arbuckle BS, Rossi C, Mattiangeli V, Lawlor PA, Mashkour M, Sauer E, Lesur J, Atici L, Cevdet CM and Bradley DG (2022) A novel lineage of the Capra genus discovered in the Taurus Mountains of Turkey using ancient genomics. bioRxiv, 2022.04.08.487619, ver. 5 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2022.04.08.487619 | A novel lineage of the Capra genus discovered in the Taurus Mountains of Turkey using ancient genomics | Kevin G. Daly, Benjamin S. Arbuckle, Conor Rossi, Valeria Mattiangeli, Phoebe A. Lawlor, Marjan Mashkour, Eberhard Sauer, Joséphine Lesur, Levent Atici, Cevdet Merih Erek, Daniel G. Bradley | <p>Direkli Cave, located in the Taurus Mountains of southern Turkey, was occupied by Late Epipaleolithic hunters-gatherers for the seasonal hunting and processing of game including large numbers of wild goats. We report genomic data from new and p... | | Evolutionary genomics, Population genomics, Vertebrates | Laura Botigué | 2022-04-15 12:05:47 | ||
20 Jul 2021
Genetic mapping of sex and self-incompatibility determinants in the androdioecious plant Phillyrea angustifoliaIdentification of distinct YX-like loci for sex determination and self-incompatibility in an androdioecious shrubRecommended by Tatiana Giraud and Ricardo C. Rodríguez de la Vega based on reviews by 2 anonymous reviewersA wide variety of systems have evolved to control mating compatibility in sexual organisms. Their genetic determinism and the factors controlling their evolution represent fascinating questions in evolutionary biology and genomics. The plant Phillyrea angustifolia (Oleaeceae family) represents an exciting model organism, as it displays two distinct and rare mating compatibility systems [1]: 1) males and hermaphrodites co-occur in populations of this shrub (a rare system called androdioecy), while the evolution and maintenance of purely hermaphroditic plants or mixtures of females and hermaphrodites (a system called gynodioecy) are easier to explain [2]; 2) a homomorphic diallelic self-incompatibility system acts in hermaphrodites, while such systems are usually multi-allelic, as rare alleles are advantageous, being compatible with all other alleles. Previous analyses of crosses brought some interesting answers to these puzzles, showing that males benefit from the ability to mate with all hermaphrodites regardless of their allele at the self-incompatibility system, and suggesting that both sex and self incompatibility are determined by XY-like genetic systems, i.e. with each a dominant allele; homozygotes for a single allele and heterozygotes therefore co-occur in natural populations at both sex and self-incompatibility loci [3]. Here, Carré et al. used genotyping-by-sequencing to build a genome linkage map of P. angustifolia [4]. The elegant and original use of a probabilistic model of segregating alleles (implemented in the SEX-DETector method) allowed to identify both the sex and self-incompatibility loci [4], while this tool was initially developed for detecting sex-linked genes in species with strictly separated sexes (dioecy) [5]. Carré et al. [4] confirmed that the sex and self-incompatibility loci are located in two distinct linkage groups and correspond to XY-like systems. A comparison with the genome of the closely related Olive tree indicated that their self-incompatibility systems were homologous. Such a XY-like system represents a rare genetic determination mechanism for self-incompatibility and has also been recently found to control mating types in oomycetes [6]. This study [4] paves the way for identifying the genes controlling the sex and self-incompatibility phenotypes and for understanding why and how self-incompatibility is only expressed in hermaphrodites and not in males. It will also be fascinating to study more finely the degree and extent of genomic differentiation at these two loci and to assess whether recombination suppression has extended stepwise away from the sex and self-incompatibility loci, as can be expected under some hypotheses, such as the sheltering of deleterious alleles near permanently heterozygous alleles [7]. Furthermore, the co-occurrence in P. angustifolia of sex and mating types can contribute to our understanding of the factor controlling their evolution [8]. References [1] Saumitou-Laprade P, Vernet P, Vassiliadis C, Hoareau Y, Magny G de, Dommée B, Lepart J (2010) A Self-Incompatibility System Explains High Male Frequencies in an Androdioecious Plant. Science, 327, 1648–1650. https://doi.org/10.1126/science.1186687 [2] Pannell JR, Voillemot M (2015) Plant Mating Systems: Female Sterility in the Driver’s Seat. Current Biology, 25, R511–R514. https://doi.org/10.1016/j.cub.2015.04.044 [3] Billiard S, Husse L, Lepercq P, Godé C, Bourceaux A, Lepart J, Vernet P, Saumitou-Laprade P (2015) Selfish male-determining element favors the transition from hermaphroditism to androdioecy. Evolution, 69, 683–693. https://doi.org/10.1111/evo.12613 [4] Carre A, Gallina S, Santoni S, Vernet P, Gode C, Castric V, Saumitou-Laprade P (2021) Genetic mapping of sex and self-incompatibility determinants in the androdioecious plant Phillyrea angustifolia. bioRxiv, 2021.04.15.439943, ver. 7 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2021.04.15.439943 [5] Muyle A, Käfer J, Zemp N, Mousset S, Picard F, Marais GA (2016) SEX-DETector: A Probabilistic Approach to Study Sex Chromosomes in Non-Model Organisms. Genome Biology and Evolution, 8, 2530–2543. https://doi.org/10.1093/gbe/evw172 [6] Dussert Y, Legrand L, Mazet ID, Couture C, Piron M-C, Serre R-F, Bouchez O, Mestre P, Toffolatti SL, Giraud T, Delmotte F (2020) Identification of the First Oomycete Mating-type Locus Sequence in the Grapevine Downy Mildew Pathogen, Plasmopara viticola. Current Biology, 30, 3897-3907.e4. https://doi.org/10.1016/j.cub.2020.07.057 [7] Jay P, Tezenas E, Giraud T (2021) A deleterious mutation-sheltering theory for the evolution of sex chromosomes and supergenes. bioRxiv, 2021.05.17.444504. https://doi.org/10.1101/2021.05.17.444504 [8] Billiard S, López-Villavicencio M, Devier B, Hood ME, Fairhead C, Giraud T (2011) Having sex, yes, but with whom? Inferences from fungi on the evolution of anisogamy and mating types. Biological Reviews, 86, 421–442. https://doi.org/10.1111/j.1469-185X.2010.00153.x | Genetic mapping of sex and self-incompatibility determinants in the androdioecious plant Phillyrea angustifolia | Amelie Carre, Sophie Gallina, Sylvain Santoni, Philippe Vernet, Cecile Gode, Vincent Castric, Pierre Saumitou-Laprade | <p style="text-align: justify;">The diversity of mating and sexual systems in angiosperms is spectacular, but the factors driving their evolution remain poorly understood. In plants of the Oleaceae family, an unusual self-incompatibility (SI) syst... | | Evolutionary genomics, Plants | Tatiana Giraud | 2021-05-04 10:37:26 | ||
11 Mar 2021

Gut microbial ecology of Xenopus tadpoles across life stagesA comprehensive look at Xenopus gut microbiota: effects of feed, developmental stages and parental transmissionRecommended by Wirulda Pootakham based on reviews by Vanessa Marcelino and 1 anonymous reviewerIt is well established that the gut microbiota play an important role in the overall health of their hosts (Jandhyala et al. 2015). To date, there are still a limited number of studies on the complex microbial communites inhabiting vertebrate digestive systems, especially the ones that also explored the functional diversity of the microbial community (Bletz et al. 2016). This preprint by Scalvenzi et al. (2021) reports a comprehensive study on the phylogenetic and metabolic profiles of the Xenopus gut microbiota. The author describes significant changes in the gut microbiome communities at different developmental stages and demonstrates different microbial community composition across organs. In addition, the study also investigates the impact of diet on the Xenopus tadpole gut microbiome communities as well as how the bacterial communities are transmitted from parents to the next generation. This is one of the first studies that addresses the interactions between gut bacteria and tadpoles during the development. The authors observe the dynamics of gut microbiome communities during tadpole growth and metamorphosis. They also explore host-gut microbial community metabolic interactions and demostrate the capacity of the microbiome to complement the metabolic pathways of the Xenopus genome. Although this study is limited by the use of Xenopus tadpoles in a laboratory, which are probably different from those in nature, I believe it still provides important and valuable information for the research community working on vertebrate’s microbiota and their interaction with the host. References Bletz et al. (2016). Amphibian gut microbiota shifts differentially in community structure but converges on habitat-specific predicted functions. Nature Communications, 7(1), 1-12. doi: https://doi.org/10.1038/ncomms13699 Jandhyala, S. M., Talukdar, R., Subramanyam, C., Vuyyuru, H., Sasikala, M., & Reddy, D. N. (2015). Role of the normal gut microbiota. World journal of gastroenterology: WJG, 21(29), 8787. doi: https://dx.doi.org/10.3748%2Fwjg.v21.i29.8787 Scalvenzi, T., Clavereau, I., Bourge, M. & Pollet, N. (2021) Gut microbial ecology of Xenopus tadpoles across life stages. bioRxiv, 2020.05.25.110734, ver. 4 peer-reviewed and recommended by Peer community in Geonmics. https://doi.org/10.1101/2020.05.25.110734 | Gut microbial ecology of Xenopus tadpoles across life stages | Thibault Scalvenzi, Isabelle Clavereau, Mickael Bourge, Nicolas Pollet | <p><strong>Background</strong> The microorganism world living in amphibians is still largely under-represented and under-studied in the literature. Among anuran amphibians, African clawed frogs of the Xenopus genus stand as well-characterized mode... | | Evolutionary genomics, Metagenomics, Vertebrates | Wirulda Pootakham | 2020-05-25 14:01:19 | ||
23 Mar 2022
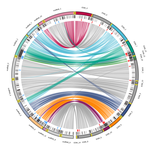
Chromosomal rearrangements with stable repertoires of genes and transposable elements in an invasive forest-pathogenic fungusComparative genomics in the chestnut blight fungus Cryphonectria parasitica reveals large chromosomal rearrangements and a stable genome organizationRecommended by Sebastien Duplessis based on reviews by Benjamin Schwessinger and 1 anonymous reviewerAbout twenty-five years after the sequencing of the first fungal genome and a dozen years after the first plant pathogenic fungi genomes were sequenced, unprecedented international efforts have led to an impressive collection of genomes available for the community of mycologists in international databases (Goffeau et al. 1996, Dean et al. 2005; Spatafora et al. 2017). For instance, to date, the Joint Genome Institute Mycocosm database has collected more than 2,100 fungal genomes over the fungal tree of life (https://mycocosm.jgi.doe.gov). Such resources are paving the way for comparative genomics, population genomics and phylogenomics to address a large panel of questions regarding the biology and the ecology of fungal species. Early on, population genomics applied to pathogenic fungi revealed a great diversity of genome content and organization and a wide variety of variants and rearrangements (Raffaele and Kamoun 2012, Hartmann 2022). Such plasticity raises questions about how to choose a representative genome to serve as an ideal reference to address pertinent biological questions. Cryphonectria parasitica is a fungal pathogen that is infamous for the devastation of chestnut forests in North America after its accidental introduction more than a century ago (Anagnostakis 1987). Since then, it has been a quarantine species under surveillance in various parts of the world. As for other fungi causing diseases on forest trees, the study of adaptation to its host in the forest ecosystem and of its reproduction and dissemination modes is more complex than for crop-targeting pathogens. A first reference genome was published in 2020 for the chestnut blight fungus C. parasitica strain EP155 in the frame of an international project with the DOE JGI (Crouch et al. 2020). Another genome was then sequenced from the French isolate YVO003, which showed a few differences in the assembly suggesting possible rearrangements (Demené et al. 2019). Here the sequencing of a third isolate ESM015 from the native area of C. parasitica in Japan allows to draw broader comparative analysis and particularly to compare between native and introduced isolates (Demené et al. 2022). Demené and collaborators report on a new genome sequence using up-to-date long-read sequencing technologies and they provide an improved genome assembly. Comparison with previously published C. parasitica genomes did not reveal dramatic changes in the overall chromosomal landscapes, but large rearrangements could be spotted. Despite these rearrangements, the genome content and organization – i.e. genes and repeats – remain stable, with a limited number of genes gains and losses. As in any fungal plant pathogen genome, the repertoire of candidate effectors predicted among secreted proteins was more particularly scrutinized. Such effector genes have previously been reported in other pathogens in repeat-enriched plastic genomic regions with accelerated evolutionary rates under the pressure of the host immune system (Raffaele and Kamoun 2012). Demené and collaborators established a list of priority candidate effectors in the C. parasitica gene catalog likely involved in the interaction with the host plant which will require more attention in future functional studies. Six major inter-chromosomal translocations were detected and are likely associated with double break strands repairs. The authors speculate on the possible effects that these translocations may have on gene organization and expression regulation leading to dramatic phenotypic changes in relation to introduction and invasion in new continents and the impact regarding sexual reproduction in this fungus (Demené et al. 2022). I recommend this article not only because it is providing an improved assembly of a reference genome for C. parasitica, but also because it adds diversity in terms of genome references availability, with a third high-quality assembly. Such an effort in the tree pathology community for a pathogen under surveillance is of particular importance for future progress in post-genomic analysis, e.g. in further genomic population studies (Hartmann 2022). References Anagnostakis SL (1987) Chestnut Blight: The Classical Problem of an Introduced Pathogen. Mycologia, 79, 23–37. https://doi.org/10.2307/3807741 Crouch JA, Dawe A, Aerts A, Barry K, Churchill ACL, Grimwood J, Hillman BI, Milgroom MG, Pangilinan J, Smith M, Salamov A, Schmutz J, Yadav JS, Grigoriev IV, Nuss DL (2020) Genome Sequence of the Chestnut Blight Fungus Cryphonectria parasitica EP155: A Fundamental Resource for an Archetypical Invasive Plant Pathogen. Phytopathology®, 110, 1180–1188. https://doi.org/10.1094/PHYTO-12-19-0478-A Dean RA, Talbot NJ, Ebbole DJ, Farman ML, Mitchell TK, Orbach MJ, Thon M, Kulkarni R, Xu J-R, Pan H, Read ND, Lee Y-H, Carbone I, Brown D, Oh YY, Donofrio N, Jeong JS, Soanes DM, Djonovic S, Kolomiets E, Rehmeyer C, Li W, Harding M, Kim S, Lebrun M-H, Bohnert H, Coughlan S, Butler J, Calvo S, Ma L-J, Nicol R, Purcell S, Nusbaum C, Galagan JE, Birren BW (2005) The genome sequence of the rice blast fungus Magnaporthe grisea. Nature, 434, 980–986. https://doi.org/10.1038/nature03449 Demené A., Laurent B., Cros-Arteil S., Boury C. and Dutech C. 2022. Chromosomal rearrangements with stable repertoires of genes and transposable elements in an invasive forest-pathogenic fungus. bioRxiv, 2021.03.09.434572, ver.6 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2021.03.09.434572 Goffeau A, Barrell BG, Bussey H, Davis RW, Dujon B, Feldmann H, Galibert F, Hoheisel JD, Jacq C, Johnston M, Louis EJ, Mewes HW, Murakami Y, Philippsen P, Tettelin H, Oliver SG (1996) Life with 6000 Genes. Science, 274, 546–567. https://doi.org/10.1126/science.274.5287.546 Hartmann FE (2022) Using structural variants to understand the ecological and evolutionary dynamics of fungal plant pathogens. New Phytologist, 234, 43–49. https://doi.org/10.1111/nph.17907 Raffaele S, Kamoun S (2012) Genome evolution in filamentous plant pathogens: why bigger can be better. Nature Reviews Microbiology, 10, 417–430. https://doi.org/10.1038/nrmicro2790 Spatafora JW, Aime MC, Grigoriev IV, Martin F, Stajich JE, Blackwell M (2017) The Fungal Tree of Life: from Molecular Systematics to Genome-Scale Phylogenies. Microbiology Spectrum, 5, 5.5.03. https://doi.org/10.1128/microbiolspec.FUNK-0053-2016 | Chromosomal rearrangements with stable repertoires of genes and transposable elements in an invasive forest-pathogenic fungus | Arthur Demene, Benoit Laurent, Sandrine Cros-Arteil, Christophe Boury, Cyril Dutech | <p style="text-align: justify;">Chromosomal rearrangements have been largely described among eukaryotes, and may have important consequences on evolution of species. High genome plasticity has been often reported in Fungi, which may explain their ... | | Evolutionary genomics, Fungi | Sebastien Duplessis | 2021-03-12 14:18:20 |




