
Latest recommendations

| Id | Title | Authors▲ | Abstract | Picture | Thematic fields | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
08 Apr 2022

POSTPRINT
Phylogenetics in the Genomic Era“Phylogenetics in the Genomic Era” brings together experts in the field to present a comprehensive synthesisRecommended by Robert Waterhouse and Karen MeusemannE-book: Phylogenetics in the Genomic Era (Scornavacca et al. 2021) This book was not peer-reviewed by PCI Genomics. It has undergone an internal review by the editors. Accurate reconstructions of the relationships amongst species and the genes encoded in their genomes are an essential foundation for almost all evolutionary inferences emerging from downstream analyses. Molecular phylogenetics has developed as a field over many decades to build suites of models and methods to reconstruct reliable trees that explain, support, or refute such inferences. The genomic era has brought new challenges and opportunities to the field, opening up new areas of research and algorithm development to take advantage of the accumulating large-scale data. Such ‘big-data’ phylogenetics has come to be known as phylogenomics, which broadly aims to connect molecular and evolutionary biology research to address questions centred on relationships amongst taxa, mechanisms of molecular evolution, and the biological functions of genes and other genomic elements. This book brings together experts in the field to present a comprehensive synthesis of Phylogenetics in the Genomic Era, covering key conceptual and methodological aspects of how to build accurate phylogenies and how to apply them in molecular and evolutionary research. The paragraphs below briefly summarise the five constituent parts of the book, highlighting the key concepts, methods, and applications that each part addresses. Being organised in an accessible style, while presenting details to provide depth where necessary, and including guides describing real-world examples of major phylogenomic tools, this collection represents an invaluable resource, particularly for students and newcomers to the field of phylogenomics. Part 1: Phylogenetic analyses in the genomic era Modelling how sequences evolve is a fundamental cornerstone of phylogenetic reconstructions. This part of the book introduces the reader to phylogenetic inference methods and algorithmic optimisations in the contexts of Markov, Maximum Likelihood, and Bayesian models of sequence evolution. The main concepts and theoretical considerations are mapped out for probabilistic Markov models, efficient tree building with Maximum Likelihood methods, and the flexibility and robustness of Bayesian approaches. These are supported with practical examples of phylogenomic applications using the popular tools RAxML and PhyloBayes. By considering theoretical, algorithmic, and practical aspects, these chapters provide readers with a holistic overview of the challenges and recent advances in developing scalable phylogenetic analyses in the genomic era. Part 2: Data quality, model adequacy This part focuses on the importance of considering the appropriateness of the evolutionary models used and the accuracy of the underlying molecular and genomic data. Both these aspects can profoundly affect the results when applying current phylogenomic methods to make inferences about complex biological and evolutionary processes. A clear example is presented for methods for building multiple sequence alignments and subsequent filtering approaches that can greatly impact phylogeny inference. The importance of error detection in (meta)barcode sequencing data is also highlighted, with solutions offered by the MACSE_BARCODE pipeline for accurate taxonomic assignments. Orthology datasets are essential markers for phylogenomic inferences, but the overview of concepts and methods presented shows that they too face challenges with respect to model selection and data quality. Finally, an innovative approach using ancestral gene order reconstructions provides new perspectives on how to assess gene tree accuracy for phylogenomic analyses. By emphasising through examples the importance of using appropriate evolutionary models and assessing input data quality, these chapters alert readers to key limitations that the field as a whole strives to address. Part 3: Resolving phylogenomic conflicts Conflicting phylogenetic signals are commonplace and may derive from statistical or systematic bias. This part of the book addresses possible causes of conflict, discordance between gene trees and species trees and how processes that lead to such conflicts can be described by phylogenetic models. Furthermore, it provides an overview of various models and methods with examples in phylogenomics including their pros and cons. Outlined in detail is the multispecies coalescent model (MSC) and its applications in phylogenomics. An interesting aspect is that different phylogenetic signals leading to conflict are in fact a key source of information rather than a problem that can – and should – be used to point to events like introgression or hybridisation, highlighting possible future trends in this research area. Last but not least, this part of the book also addresses inferring species trees by concatenating single multiple sequence alignments (gene alignments) versus inferring the species tree based on ensembles of single gene trees pointing out advantages and disadvantages of both approaches. As an important take home message from these chapters, it is recommended to be flexible and identify the most appropriate approach for each dataset to be analysed since this may tremendously differ depending on the dataset, setting, taxa, and phylogenetic level addressed by the researcher. Part 4: Functional evolutionary genomics In this part of the book the focus shifts to functional considerations of phylogenomics approaches both in terms of molecular evolution and adaptation and with respect to gene expression. The utility of multi-species analysis is clearly presented in the context of annotating functional genomic elements through quantifying evolutionary constraint and protein-coding potential. An historical perspective on characterising rates of change highlights how phylogenomic datasets help to understand the modes of molecular evolution across the genome, over time, and between lineages. These are contextualised with respect to the specific aim of detecting signatures of adaptation from protein-coding DNA alignments using the example of the MutSelDP-ω∗ model. This is extended with the presentation of the generally rare case of adaptive sequence convergence, where consideration of appropriate models and knowledge of gene functions and phenotypic effects are needed. Constrained or relaxed, selection pressures on sequence or copy-number affect genomic elements in different ways, making the very concept of function difficult to pin down despite it being fundamental to relate the genome to the phenotype and organismal fitness. Here gene expression provides a measurable intermediate, for which the Expression Comparison tool from the Bgee suite allows exploration of expression patterns across multiple animal species taking into account anatomical homology. Overall, phylogenomics applications in functional evolutionary genomics build on a rich theoretical history from molecular analyses where integration with knowledge of gene functions is challenging but critical. Part 5: Phylogenomic applications Rather than attempting to review the full extent of applications linked to phylogenomics, this part of the book focuses on providing detailed specific insights into selected examples and methods concerning i) estimating divergence times, and ii) species delimitation in the era of ‘omics’ data. With respect to estimating divergence times, an exemplary overview is provided for fossil data recovered from geological records, either using fossil data as calibration points with an extant-species-inferred phylogeny, or using a fossilised birth-death process as a mechanistic model that accounts for lineage diversification. Included is a tutorial for a joint approach to infer phylogenies and estimate divergence times using the RevBayes software with various models implemented for different applications and datasets incorporating molecular and morphological data. An interesting excursion is outlined focusing on timescale estimates with respect to viral evolution introducing BEAGLE, a high-performance likelihood-calculation platform that can be used on multi-core systems. As a second major subject, species delimitation is addressed since currently the increasing amount of available genomic data enables extensive inferences, for instance about the degree of genetic isolation among species and ancient and recent introgression events. Describing the history of molecular species delimitation up to the current genomic era and presenting widely used computational methods incorporating single- and multi-locus genomic data, pros and cons are addressed. Finally, a proposal for a new method for delimiting species based on empirical criteria is outlined. In the closing chapter of this part of the book, BPP (Bayesian Markov chain Monte Carlo program) for analysing multi-locus sequence data under the multispecies coalescent (MSC) model with and without introgression is introduced, including a tutorial. These examples together provide accessible details on key conceptual and methodological aspects related to the application of phylogenetics in the genomic era. References Scornavacca C, Delsuc F, Galtier N (2021) Phylogenetics in the Genomic Era. https://hal.inria.fr/PGE/ | Phylogenetics in the Genomic Era | Céline Scornavacca, Frédéric Delsuc, Nicolas Galtier | <p style="text-align: justify;">Molecular phylogenetics was born in the middle of the 20th century, when the advent of protein and DNA sequencing offered a novel way to study the evolutionary relationships between living organisms. The first 50 ye... | | Bacteria and archaea, Bioinformatics, Evolutionary genomics, Functional genomics, Fungi, Plants, Population genomics, Vertebrates, Viruses and transposable elements | Robert Waterhouse | 2022-03-15 17:43:52 | ||
07 Aug 2023
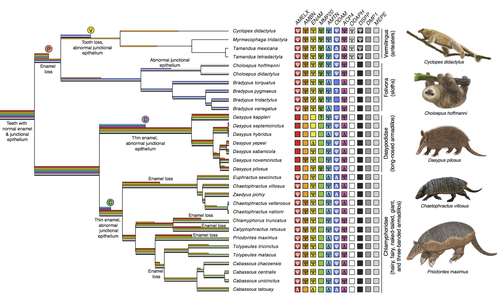
Genomic data suggest parallel dental vestigialization within the xenarthran radiationWhat does dental gene decay tell us about the regressive evolution of teeth in South American mammals?Recommended by Didier Casane based on reviews by Juan C. Opazo, Régis Debruyne and Nicolas PolletA group of mammals, Xenathra, evolved and diversified in South America during its long period of isolation in the early to mid Cenozoic era. More recently, as a result of the Great Faunal Interchange between South America and North America, many xenarthran species went extinct. The thirty-one extant species belong to three groups: armadillos, sloths and anteaters. They share dental degeneration. However, the level of degeneration is variable. Anteaters entirely lack teeth, sloths have intermediately regressed teeth and most armadillos have a toothless premaxilla, as well as peg-like, single-rooted teeth that lack enamel in adult animals (Vizcaíno 2009). This diversity raises a number of questions about the evolution of dentition in these mammals. Unfortunately, the fossil record is too poor to provide refined information on the different stages of regressive evolution in these clades. In such cases, the identification of loss-of-function mutations and/or relaxed selection in genes related to a character regression can be very informative (Emerling and Springer 2014; Meredith et al. 2014; Policarpo et al. 2021). Indeed, shared and unique pseudogenes/relaxed selection can tell us to what extent regression has occurred in common ancestors and whether some changes are lineage-specific. In addition, the distribution of pseudogenes/relaxed selection on the branches of a phylogenetic tree is related to the evolutionary processes involved. A much higher density of pseudogenes in the most internal branches indicates that degeneration took place early and over a short period of time, consistent with selection against the presence of the morphological character with which they are associated, while pseudogenes distributed evenly in many internal and external branches suggest a more gradual process over many millions of years, in line with relaxed selection and fixation of loss-of-function mutations by genetic drift. In this paper (Emerling et al. 2023), the authors examined the dynamics of decay of 11 dental genes that may parallel teeth regression. The analyses of the data reported in this paper clearly point to xenarthran teeth having repeatedly regressed in parallel in the three clades. In fact, no loss-of-function mutation is shared by all species examined. However, more genes should be studied to confirm the hypothesis that the common ancestor of extant xenarthrans had normal dentition. There are distinct patterns of gene loss in different lineages that are associated with the variation in dentition observed across the clades. These patterns of gene loss suggest that regressive evolution took place both gradually and in relatively rapid, discrete phases during the diversification of xenarthrans. This study underscores the utility of using pseudogenes to reconstruct evolutionary history of morphological characters when fossils are sparse. References Emerling CA, Gibb GC, Tilak M-K, Hughes JJ, Kuch M, Duggan AT, Poinar HN, Nachman MW, Delsuc F. 2023. Genomic data suggest parallel dental vestigialization within the xenarthran radiation. bioRxiv, 2022.12.09.519446, ver 2, peer-reviewed and recommended by PCI Genomics. https://doi.org/10.1101/2022.12.09.519446 Emerling CA, Springer MS. 2014. Eyes underground: Regression of visual protein networks in subterranean mammals. Molecular Phylogenetics and Evolution 78: 260-270. https://doi.org/10.1016/j.ympev.2014.05.016 Meredith RW, Zhang G, Gilbert MTP, Jarvis ED, Springer MS. 2014. Evidence for a single loss of mineralized teeth in the common avian ancestor. Science 346: 1254390. https://doi.org/10.1126/science.1254390 Policarpo M, Fumey J, Lafargeas P, Naquin D, Thermes C, Naville M, Dechaud C, Volff J-N, Cabau C, Klopp C, et al. 2021. Contrasting gene decay in subterranean vertebrates: insights from cavefishes and fossorial mammals. Molecular Biology and Evolution 38: 589-605. https://doi.org/10.1093/molbev/msaa249 Vizcaíno SF. 2009. The teeth of the “toothless”: novelties and key innovations in the evolution of xenarthrans (Mammalia, Xenarthra). Paleobiology 35: 343-366. https://doi.org/10.1666/0094-8373-35.3.343 | Genomic data suggest parallel dental vestigialization within the xenarthran radiation | Christopher A Emerling, Gillian C Gibb, Marie-Ka Tilak, Jonathan J Hughes, Melanie Kuch, Ana T Duggan, Hendrik N Poinar, Michael W Nachman, Frederic Delsuc | <p style="text-align: justify;">The recent influx of genomic data has provided greater insights into the molecular basis for regressive evolution, or vestigialization, through gene loss and pseudogenization. As such, the analysis of gene degradati... | | Evolutionary genomics, Vertebrates | Didier Casane | 2022-12-12 16:01:57 | ||
02 Jun 2023
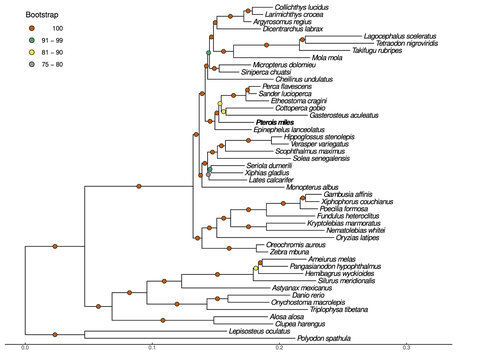
Near-chromosome level genome assembly of devil firefish, Pterois milesThe genome of a dangerous invader (fish) beautyRecommended by Iker Irisarri based on reviews by Maria Recuerda and 1 anonymous reviewer based on reviews by Maria Recuerda and 1 anonymous reviewer
High-quality genomes are currently being generated at an unprecedented speed powered by long-read sequencing technologies. However, sequencing effort is concentrated unequally across the tree of life and several key evolutionary and ecological groups remain largely unexplored. So is the case for fish species of the family Scorpaenidae (Perciformes). Kitsoulis et al. present the genome of the devil firefish, Pterois miles (1). Following current best practices, the assembly relies largely on Oxford Nanopore long reads, aided by Illumina short reads for polishing to increase the per-base accuracy. PacBio’s IsoSeq was used to sequence RNA from a variety of tissues as direct evidence for annotating genes. The reconstructed genome is 902 Mb in size and has high contiguity (N50=14.5 Mb; 660 scaffolds, 90% of the genome covered by the 83 longest scaffolds) and completeness (98% BUSCO completeness). The new genome is used to assess the phylogenetic position of P. miles, explore gene synteny against zebrafish, look at orthogroup expansion and contraction patterns in Perciformes, as well as to investigate the evolution of toxins in scorpaenid fish (2). In addition to its value for better understanding the evolution of scorpaenid and teleost fishes, this new genome is also an important resource for monitoring its invasiveness through the Mediterranean Sea (3) and the Atlantic Ocean, in the latter case forming the invasive lionfish complex with P. volitans (4). REFERENCES 1. Kitsoulis CV, Papadogiannis V, Kristoffersen JB, Kaitetzidou E, Sterioti E, Tsigenopoulos CS, Manousaki T. (2023) Near-chromosome level genome assembly of devil firefish, Pterois miles. BioRxiv, ver. 6 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2023.01.10.523469 2. Kiriake A, Shiomi K. (2011) Some properties and cDNA cloning of proteinaceous toxins from two species of lionfish (Pterois antennata and Pterois volitans). Toxicon, 58(6-7):494–501. https://doi.org/10.1016/j.toxicon.2011.08.010 3. Katsanevakis S, et al. (2020) Un- published Mediterranean records of marine alien and cryptogenic species. BioInvasions Records, 9:165–182. https://doi.org/10.3391/bir.2020.9.2.01 4. Lyons TJ, Tuckett QM, Hill JE. (2019) Data quality and quantity for invasive species: A case study of the lionfishes. Fish and Fisheries, 20:748–759. https://doi.org/10.1111/faf.12374 | Near-chromosome level genome assembly of devil firefish, *Pterois miles* | Christos V. Kitsoulis, Vasileios Papadogiannis, Jon B. Kristoffersen, Elisavet Kaitetzidou, Aspasia Sterioti, Costas S. Tsigenopoulos, Tereza Manousaki | <p style="text-align: justify;">Devil firefish (<em>Pterois miles</em>), a member of Scorpaenidae family, is one of the most successful marine non-native species, dominating around the world, that was rapidly spread into the Mediterranean Sea, thr... | | Evolutionary genomics | Iker Irisarri | 2023-01-17 12:37:20 | ||
13 Jul 2022
Nucleosome patterns in four plant pathogenic fungi with contrasted genome structuresGenome-wide chromatin and expression datasets of various pathogenic ascomycetesRecommended by Sébastien Bloyer and Romain Koszul based on reviews by Ricardo C. Rodríguez de la Vega and 1 anonymous reviewerPlant pathogenic fungi represent serious economic threats. These organisms are rapidly adaptable, with plastic genomes containing many variable regions and evolving rapidly. It is, therefore, useful to characterize their genetic regulation in order to improve their control. One of the steps to do this is to obtain omics data that link their DNA structure and gene expression. Clairet C, Lapalu N, Simon A, Soyer JL, Viaud M, Zehraoui E, Dalmais B, Fudal I, Ponts N (2022) Nucleosome patterns in four plant pathogenic fungi with contrasted genome structures. bioRxiv, 2021.04.16.439968, ver. 4 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2021.04.16.439968 | Nucleosome patterns in four plant pathogenic fungi with contrasted genome structures | Colin Clairet, Nicolas Lapalu, Adeline Simon, Jessica L. Soyer, Muriel Viaud, Enric Zehraoui, Berengere Dalmais, Isabelle Fudal, Nadia Ponts | <p style="text-align: justify;">Fungal pathogens represent a serious threat towards agriculture, health, and environment. Control of fungal diseases on crops necessitates a global understanding of fungal pathogenicity determinants and their expres... | | Epigenomics, Fungi | Sébastien Bloyer | 2021-04-17 10:32:41 | ||
24 Jan 2024

High quality genome assembly of the brown hare (Lepus europaeus) with chromosome-level scaffoldingA high quality reference genome of the brown hareRecommended by Ed Hollox based on reviews by Merce Montoliu-Nerin and 1 anonymous reviewer
The brown hare, or European hare, Lupus europaeus, is a widespread mammal whose natural range spans western Eurasia. At the northern limit of its range, it hybridises with the mountain hare (L. timidis), and humans have introduced it into other continents. It represents a particularly interesting mammal to study for its population genetics, extensive hybridisation zones, and as an invasive species. This study (Michell et al. 2024) has generated a high-quality assembly of a genome from a brown hare from Finland using long PacBio HiFi sequencing reads and Hi-C scaffolding. The contig N50 of this new genome is 43 Mb, and completeness, assessed using BUSCO, is 96.1%. The assembly comprises 23 autosomes, and an X chromosome and Y chromosome, with many chromosomes including telomeric repeats, indicating the high level of completeness of this assembly. While the genome of the mountain hare has previously been assembled, its assembly was based on a short-read shotgun assembly, with the rabbit as a reference genome. The new high-quality brown hare genome assembly allows a direct comparison with the rabbit genome assembly. For example, the assembly addresses the karyotype difference between the hare (n=24) and the rabbit (n=22). Chromosomes 12 and 17 of the hare are equivalent to chromosome 1 of the rabbit, and chromosomes 13 and 16 of the hare are equivalent to chromosome 2 of the rabbit. The new assembly also provides a hare Y-chromosome, as the previous mountain hare genome was from a female. This new genome assembly provides an important foundation for population genetics and evolutionary studies of lagomorphs. References Michell, C., Collins, J., Laine, P. K., Fekete, Z., Tapanainen, R., Wood, J. M. D., Goffart, S., Pohjoismäki, J. L. O. (2024). High quality genome assembly of the brown hare (Lepus europaeus) with chromosome-level scaffolding. bioRxiv, ver. 3 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2023.08.29.555262 | High quality genome assembly of the brown hare (*Lepus europaeus*) with chromosome-level scaffolding | Craig Michell, Joanna Collins, Pia K. Laine, Zsofia Fekete, Riikka Tapanainen, Jonathan M. D. Wood, Steffi Goffart, Jaakko L. O. Pohjoismaki | <p style="text-align: justify;">We present here a high-quality genome assembly of the brown hare (Lepus europaeus Pallas), based on a fibroblast cell line of a male specimen from Liperi, Eastern Finland. This brown hare genome represents the first... | | ERGA Pilot, Vertebrates | Ed Hollox | 2023-10-16 20:46:39 | ||
15 Sep 2022
EukProt: A database of genome-scale predicted proteins across the diversity of eukaryotesEukProt enables reproducible Eukaryota-wide protein sequence analysesRecommended by Gavin Douglas based on reviews by 2 anonymous reviewers
Comparative genomics is a general approach for understanding how genomes differ, which can be considered from many angles. For instance, this approach can delineate how gene content varies across organisms, which can lead to novel hypotheses regarding what those organisms do. It also enables investigations into the sequence-level divergence of orthologous DNA, which can provide insight into how evolutionary forces differentially shape genome content and structure across lineages. Burki F, Roger AJ, Brown MW, Simpson AGB (2020) The New Tree of Eukaryotes. Trends in Ecology & Evolution, 35, 43–55. https://doi.org/10.1016/j.tree.2019.08.008 Richter DJ, Berney C, Strassert JFH, Poh Y-P, Herman EK, Muñoz-Gómez SA, Wideman JG, Burki F, Vargas C de (2022) EukProt: A database of genome-scale predicted proteins across the diversity of eukaryotes. bioRxiv, 2020.06.30.180687, ver. 5 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2020.06.30.180687 Wilkinson MD, Dumontier M, Aalbersberg IjJ, Appleton G, Axton M, Baak A, Blomberg N, Boiten J-W, da Silva Santos LB, Bourne PE, Bouwman J, Brookes AJ, Clark T, Crosas M, Dillo I, Dumon O, Edmunds S, Evelo CT, Finkers R, Gonzalez-Beltran A, Gray AJG, Groth P, Goble C, Grethe JS, Heringa J, ’t Hoen PAC, Hooft R, Kuhn T, Kok R, Kok J, Lusher SJ, Martone ME, Mons A, Packer AL, Persson B, Rocca-Serra P, Roos M, van Schaik R, Sansone S-A, Schultes E, Sengstag T, Slater T, Strawn G, Swertz MA, Thompson M, van der Lei J, van Mulligen E, Velterop J, Waagmeester A, Wittenburg P, Wolstencroft K, Zhao J, Mons B (2016) The FAIR Guiding Principles for scientific data management and stewardship. Scientific Data, 3, 160018. https://doi.org/10.1038/sdata.2016.18 | EukProt: A database of genome-scale predicted proteins across the diversity of eukaryotes | Daniel J. Richter, Cédric Berney, Jürgen F. H. Strassert, Yu-Ping Poh, Emily K. Herman, Sergio A. Muñoz-Gómez, Jeremy G. Wideman, Fabien Burki, Colomban de Vargas | <p style="text-align: justify;">EukProt is a database of published and publicly available predicted protein sets selected to represent the breadth of eukaryotic diversity, currently including 993 species from all major supergroups as well as orpha... | | Bioinformatics, Evolutionary genomics | Gavin Douglas | 2022-06-08 14:19:28 | ||
14 Sep 2023
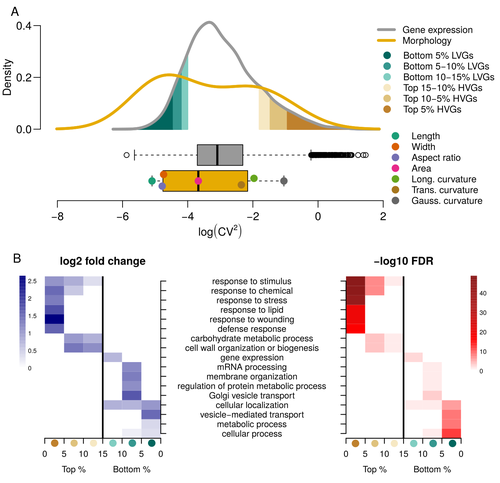
Expression of cell-wall related genes is highly variable and correlates with sepal morphologyThe same but different: How small scale hidden variations can have large effectsRecommended by Francois Sabot based on reviews by Sandra Corjito and 1 anonymous reviewer
For ages, we considered only single genes, or just a few, in order to understand the relationship between phenotype and genotype in response to environmental challenges. Recently, the use of meaningful groups of genes, e.g. gene regulatory networks, or modules of co-expression, allowed scientists to have a larger view of gene regulation. However, all these findings were based on contrasted genotypes, e.g. between wild-types and mutants, as the implicit assumption often made is that there is little transcriptomic variability within the same genotype context. Hartasànchez and collaborators (2023) decided to challenge both views: they used a single genotype instead of two, the famous A. thaliana Col0, and numerous plants, and considered whole gene networks related to sepal morphology and its variations. They used a clever approach, combining high-level phenotyping and gene expression to better understand phenomena and regulations underlying sepal morphologies. Using multiple controls, they showed that basic variations in the expression of genes related to the cell wall regulation, as well as the ones involved in chloroplast metabolism, influenced the global transcriptomic pattern observed in sepal while being in near-identical genetic background and controlling for all other experimental conditions. The paper of Hartasànchez et al. is thus a tremendous call for humility in biology, as we saw in their work that we just understand the gross machinery. However, the Devil is in the details: understanding those very small variations that may have a large influence on phenotypes, and thus on local adaptation to environmental challenges, is of great importance in these times of climatic changes. References Hartasánchez DA, Kiss A, Battu V, Soraru C, Delgado-Vaquera A, Massinon F, Brasó-Vives M, Mollier C, Martin-Magniette M-L, Boudaoud A, Monéger F. 2023. Expression of cell-wall related genes is highly variable and correlates with sepal morphology. bioRxiv, ver. 4, peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2022.04.26.489498 | Expression of cell-wall related genes is highly variable and correlates with sepal morphology | Diego A. Hartasánchez, Annamaria Kiss, Virginie Battu, Charline Soraru, Abigail Delgado-Vaquera, Florian Massinon, Marina Brasó-Vives, Corentin Mollier, Marie-Laure Martin-Magniette, Arezki Boudaoud, Françoise Monéger | <p style="text-align: justify;">Control of organ morphology is a fundamental feature of living organisms. There is, however, observable variation in organ size and shape within a given genotype. Taking the sepal of Arabidopsis as a model, we inves... | | Bioinformatics, Epigenomics, Plants | Francois Sabot | 2023-03-14 19:10:15 | ||
15 Mar 2024
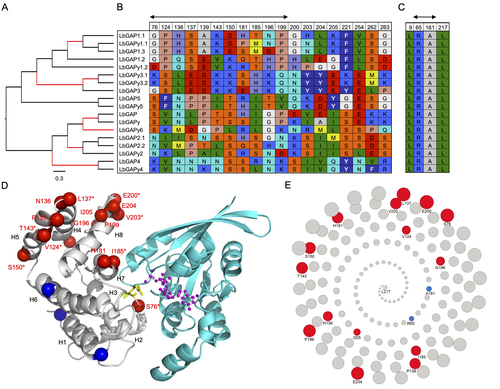
Convergent origin and accelerated evolution of vesicle-associated RhoGAP proteins in two unrelated parasitoid waspsUsing transcriptomics and proteomics to understand the expansion of a secreted poisonous armoury in parasitoid wasps genomesRecommended by Ignacio Bravo based on reviews by Inacio Azevedo and 2 anonymous reviewers
Parasitoid wasps lay their eggs inside another arthropod, whose body is physically consumed by the parasitoid larvae. Phylogenetic inference suggests that Parasitoida are monophyletic, and that this clade underwent a strong radiation shortly after branching off from the Apocrita stem, some 236 million years ago (Peters et al. 2017). The increase in taxonomic diversity during evolutionary radiations is usually concurrent with an increase in genetic/genomic diversity, and is often associated with an increase in phenotypic diversity. Gene (or genome) duplication provides the evolutionary potential for such increase of genomic diversity by neo/subfunctionalisation of one of the gene paralogs, and is often proposed to be related to evolutionary radiations (Ohno 1970; Francino 2005).
References
| Convergent origin and accelerated evolution of vesicle-associated RhoGAP proteins in two unrelated parasitoid wasps | Dominique Colinet, Fanny Cavigliasso, Matthieu Leobold, Appoline Pichon, Serge Urbach, Dominique Cazes, Marine Poullet, Maya Belghazi, Anne-Nathalie Volkoff, Jean-Michel Drezen, Jean-Luc Gatti, and Marylène Poirié | <p>Animal venoms and other protein-based secretions that perform a variety of functions, from predation to defense, are highly complex cocktails of bioactive compounds. Gene duplication, accompanied by modification of the expression and/or functio... | | Evolutionary genomics | Ignacio Bravo | 2023-06-12 11:08:31 | ||
15 Jan 2024
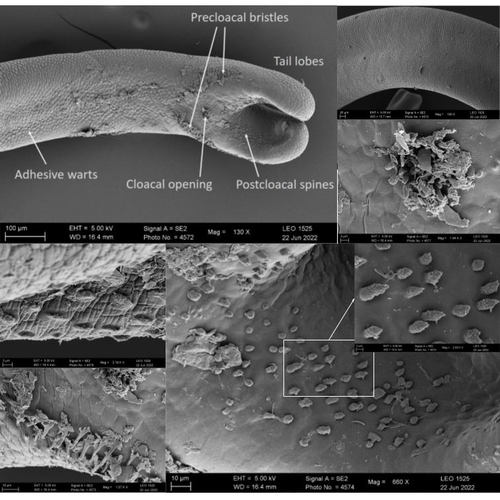
The genome sequence of the Montseny horsehair worm, Gordionus montsenyensis sp. nov., a key resource to investigate Ecdysozoa evolutionEmbarking on a novel journey in Metazoa evolution through the pioneering sequencing of a key underrepresented lineageRecommended by Juan C. Opazo based on reviews by Gonzalo Riadi and 2 anonymous reviewers
Whole genome sequences are revolutionizing our understanding across various biological fields. They not only shed light on the evolution of genetic material but also uncover the genetic basis of phenotypic diversity. The sequencing of underrepresented lineages, such as the one presented in this study, is of critical importance. It is crucial in filling significant gaps in our understanding of Metazoa evolution. Despite the wealth of genome sequences in public databases, it is crucial to acknowledge that some lineages across the Tree of Life are underrepresented or absent. This research represents a significant step towards addressing this imbalance, contributing to the collective knowledge of the global scientific community. In this genome note, as part of the European Reference Genome Atlas pilot effort to generate reference genomes for European biodiversity (Mc Cartney et al. 2023), Klara Eleftheriadi and colleagues (Eleftheriadi et al. 2023) make a significant effort to add a genome sequence of an unrepresented group in the animal Tree of Life. More specifically, they present a taxonomic description and chromosome-level genome assembly of a newly described species of horsehair worm (Gordionus montsenyensis). Their sequence methodology gave rise to an assembly of 396 scaffolds totaling 288 Mb, with an N50 value of 64.4 Mb, where 97% of this assembly is grouped into five pseudochromosomes. The nuclear genome annotation predicted 10,320 protein-coding genes, and they also assembled the circular mitochondrial genome into a 15-kilobase sequence. The selection of a species representing the phylum Nematomorpha, a group of parasitic organisms belonging to the Ecdysozoa lineage, is good, since today, there is only one publicly available genome for this animal phylum (Cunha et al. 2023). Interestingly, this article shows, among other things, that the species analyzed has lost ∼30% of the universal Metazoan genes. Efforts, like the one performed by Eleftheriadi and colleagues, are necessary to gain more insights, for example, on the evolution of this massive gene lost in this group of animals.
Cunha, T. J., de Medeiros, B. A. S, Lord, A., Sørensen, M. V., and Giribet, G. (2023). Rampant Loss of Universal Metazoan Genes Revealed by a Chromosome-Level Genome Assembly of the Parasitic Nematomorpha. Current Biology, 33 (16): 3514–21.e4. https://doi.org/10.1016/j.cub.2023.07.003 Eleftheriadi, K., Guiglielmoni, N., Salces-Ortiz, J., Vargas-Chavez, C., Martínez-Redondo, G. I., Gut, M., Flot, J.-F., Schmidt-Rhaesa, A., and Fernández, R. (2023). The Genome Sequence of the Montseny Horsehair worm, Gordionus montsenyensis sp. Nov., a Key Resource to Investigate Ecdysozoa Evolution. bioRxiv, ver. 3 peer-reviewed and recommended by Peer Community in Genomics. https://doi.org/10.1101/2023.06.26.546503 Mc Cartney, A. M., Formenti, G., Mouton, A., De Panis, D., Marins, L. S., Leitão, H. G., Diedericks, G., et al. (2023). The European Reference Genome Atlas: Piloting a Decentralised Approach to Equitable Biodiversity Genomics. bioRxiv. https://doi.org/10.1101/2023.09.25.559365 | The genome sequence of the Montseny horsehair worm, *Gordionus montsenyensis* sp. nov., a key resource to investigate Ecdysozoa evolution | Eleftheriadi Klara, Guiglielmoni Nadège, Salces-Ortiz Judit, Vargas-Chávez Carlos, Martínez-Redondo Gemma I, Gut Marta, Flot Jean François, Schmidt-Rhaesa Andreas, Fernández Rosa | <p>Nematomorpha, also known as Gordiacea or Gordian worms, are a phylum of parasitic organisms that belong to the Ecdysozoa, a clade of invertebrate animals characterized by molting. They are one of the less scientifically studied animal phyla, an... | | ERGA Pilot | Juan C. Opazo | 2023-06-29 10:31:36 | ||
09 Oct 2020
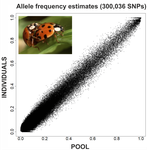
An evaluation of pool-sequencing transcriptome-based exon capture for population genomics in non-model speciesAssessing a novel sequencing-based approach for population genomics in non-model speciesRecommended by Thomas Derrien and Sebastian E. Ramos-Onsins based on reviews by Valentin Wucher and 1 anonymous reviewerDeveloping new sequencing and bioinformatic strategies for non-model species is of great interest in many applications, such as phylogenetic studies of diverse related species, but also for studies in population genomics, where a relatively large number of individuals is necessary. Different approaches have been developed and used in these last two decades, such as RAD-Seq (e.g., Miller et al. 2007), exome sequencing (e.g., Teer and Mullikin 2010) and other genome reduced representation methods that avoid the use of a good reference and well annotated genome (reviewed at Davey et al. 2011). However, population genomics studies require the analysis of numerous individuals, which makes the studies still expensive. Pooling samples was thought as an inexpensive strategy to obtain estimates of variability and other related to the frequency spectrum, thus allowing the study of variability at population level (e.g., Van Tassell et al. 2008), although the major drawback was the loss of information related to the linkage of the variants. In addition, population analysis using all these sequencing strategies require statistical and empirical validations that are not always fully performed. A number of studies aiming to obtain unbiased estimates of variability using reduced representation libraries and/or with pooled data have been performed (e.g., Futschik and Schlötterer 2010, Gautier et al. 2013, Ferretti et al. 2013, Lynch et al. 2014), as well as validation of new sequencing methods for population genetic analyses (e.g., Gautier et al. 2013, Nevado et al. 2014). Nevertheless, empirical validation using both pooled and individual experimental approaches combined with different bioinformatic methods has not been always performed. References [1] Choquet et al. (2019). Towards population genomics in non-model species with large genomes: a case study of the marine zooplankton Calanus finmarchicus. Royal Society open science, 6(2), 180608. doi: https://doi.org/10.1098/rsos.180608 | An evaluation of pool-sequencing transcriptome-based exon capture for population genomics in non-model species | Emeline Deleury, Thomas Guillemaud, Aurélie Blin & Eric Lombaert | <p>Exon capture coupled to high-throughput sequencing constitutes a cost-effective technical solution for addressing specific questions in evolutionary biology by focusing on expressed regions of the genome preferentially targeted by selection. Tr... | | Bioinformatics, Population genomics | Thomas Derrien | 2020-02-26 09:21:11 |




